生物信息学:bedGraph文件、Bed文件转、BAM文件转化
目录
引言
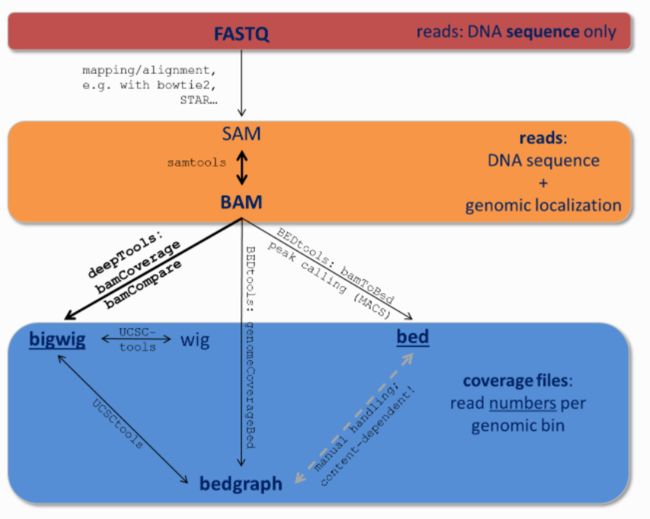
bam文件
bed文件
bedGraph文件
R语言和linux实现从bedgraph到bam的转化
从bedgraph到bed
genome.size.txt
bed转bam
引言
Bam Bed bedGraph是保存测序数据的常用格式。在GEO数据库的文件中,很多都是用bed、bedgraph储存的,如何转变为更为下层的bam呢?,这里说一下如何从bedGraph转化成bam文件。
bam文件
第一列:QNAME
进行reads比对时通常表示reads的名字,如果这条reads比对到多条序列或比对到这条序列的多个位置,相同名字会出现多次。如果是pair-end reads,相同名字会出现2次,分别表示来自于R1文件的reads和R2文件的reads,如果其matepair reads也比对2个位置,也会出现2次,则相同名字共出现4次,如果一条reads也比对2个位置,则其matepair比对1个位置,则共出现3次,如果其matepair reads没有比对上序列也会出现1次(第三列显示“*”),所以pair-end测序,R1文件和R2文件同时mapping,相同reads的id最少出现2次。
第二列:FLAG
数值结果如下:
1(1)该read是成对的paired reads中的一个
2(10)paired reads中每个都正确比对到参考序列上
4(100)该read没比对到参考序列上
8(1000)与该read成对的matepair read没有比对到参考序列上
16(10000)该read其反向互补序列能够比对到参考序列
32(100000)与该read成对的matepair read其反向互补序列能够比对到参考序列
64(1000000)在paired reads中,该read是与参考序列比对的第一条
128(10000000)在paired reads中,该read是与参考序列比对的第二条
256(100000000)该read是次优的比对结果
512(1000000000)该read没有通过质量控制
1024(10000000000)由于PCR或测序错误产生的重复reads
2048(100000000000)补充匹配的read
具体的flag值的解释,可以参考samtools软件提供的结果
samtools(Version: 1.3.1)
其中的samtools flags用法可提供flag值的查找结果
About: Convert between textual and numeric flag representation
Usage: samtools flags INT|STR[,…]
例如:
samtools flags 10
0xa 10 PROPER_PAIR,MUNMAP(10=2+8)
samtools flags 12
0xc 12 UNMAP,MUNMAP(12=4+8)
具体的flag值的解释,也可参考如下网站:https://broadinstitute.github.io/picard/explain-flags.html
或者在必应当中搜索flag sam点击Explain SAM Flags-GitHub Pages进入该网页,也可以输入组合flag数值会出现所存在的意思
第三列:RNAME
表示read比对的那条序列的序列名称(名称与头部的@SQ相对应),如果这列是“*”,可以认为这条read没有比对上的序列,则这一行的第四,五,八,九 列是“0”,第六,七列与该列是相同的表示方法
第四列:POS
表示read比对到RNAME这条序列的最左边的位置,如果该read能够完全比对到这条序列(CIGAR string为M)则这个位置是read的第一个碱基比对的位置,如果该read的反向互补序列比对到这条序列,则这个位置是read的反向互补序列的第一个碱基比对的位置,所以无论该read是正向比对到该序列,或是其反向互补序列比对到该序列,比对结果均是最左端的比对位置
第五列:MAPQ
表示为mapping的质量值,mapping Quality, It equals -10log10Pr{mapping position is wrong}, rounded to the nearest integer, A value 255 indicates that the mapping quality is not available. 该值的计算方法是mapping的错误率的-10log10值,之后四舍五入得到的整数,如果值为255表示mapping值是不可用的,如果是unmapped read则MAPQ为0,一般在使用bwa mem或bwa aln(bwa 0.7.12-r1039版本)生成的sam文件,第五列为60表示mapping率最高,一般结果是这一列的数值是从0到60,且0和60这两个数字出现次数最多
第六列:CIGAR
CIGAR string,可以理解为reads mapping到第三列序列的mapping状态,
对于mapping状态可分为以下几类:
M:alignment match (can be a sequence match or mismatch)
表示read可mapping到第三列的序列上,则read的碱基序列与第三列的序列碱基相同,表示正常的mapping结果,M表示完全匹配,但是无论reads与序列的正确匹配或是错误匹配该位置都显示为M
I:insertion to the reference
表示read的碱基序列相对于第三列的RNAME序列,有碱基的插入
D:deletion from the reference
表示read的碱基序列相对于第三列的RNAME序列,有碱基的删除
N:skipped region from the reference
表示可变剪接位置
P:padding (silent deletion from padded reference)
S:soft clipping (clipped sequences present in SEQ)
H:hard clipping (clipped sequences NOT present in SEQ)
clipped均表示一条read的序列被分开,之所以被分开,是因为read的一部分序列能匹配到第三列的RNAME序列上,而被分开的那部分不能匹配到RNAME序列上。
"="表示正确匹配到序列上
"X"表示错误匹配到序列上
而H只出现在一条read的前端或末端,但不会出现在中间,S一般会和H成对出现,当有H出现时,一定会有一个与之对应的S出现
例如:
162M89S
162H89M
149M102S
149H102M
40S211M
20M1D20M211H
S可以单独出现,而H必须有与之对应的S出现时才可能出现,不可在相同第一列的情况下单独出现
N:如果是mRNA-to-genome,N出现的位置代表内含子,其它比对形式出现N时则没有具体解释
M/I/S/=/X:这些数值的加和等于第10列SEQ的长度
第七列:MRNM
这条reads第二次比对的位置,在利用bwa mem产生sam文件时,如果该列是“”而
第3列RNAME不是“”则表示该reads比对到第3列显示序列名的序列上,而没有比对到其他位置,在利用bwa aln及bwa sampe比对生成的sam文件,如果和上述情况相同,则第7列为“=”,上述情况均表示该reads只比对到这一个位置
如果第3列RNAME和第7列MRNM都为“*”,则说明这条reads没有匹配上的序列,如果这条reads匹配两个序列,则第一个序列的名称出现在第3列,而第二个序列的名称出现在第7列
第八列:MPOS
该列表示与该reads对应的mate pair reads的比对位置,如果这对pair-end reads比对到同一条reference序列上,在sam文件中reads的id出现2次,Read1比对的第4列等于Read2比对的第8列。同样Read1比对的第8列等于Read2比对的第4列。例如:
第1列(Read id)····第4列(Read1比对位置)····第8列(mate-pair reads比对位置)
22699:1759····124057649····124057667
22699:1759····124057667····124057649
相同的reads id一个来自Read1文件,一个来自Read2文件,第4列和第8列是对应的
第九列:ISIZE
TLEN:signed observed Template LENgth (可以理解为文库插入片段长度)
如果R1端的read和R2端的read能够mapping到同一条Reference序列上(即第三列RNAME相同),则该列的值表示第8列减去第4列加上第6列的值,R1端和R2端相同id的reads其第九列值相同,但该值为一正一负,R1文件的reads和R2文件的reads,相同id的reads要相对来看。在进行该第列值的计算时,如果取第6列的数值,一定要取出现M的值,S或H的值不能取。
the unisgned observed template length equals the number of base from the leftmost mapped base to the rightmost mappedbase. Theleftmost segment has a plus sign and the rightmost has a minus sign
bed文件
bed文件一般代表区域信息,如表示peak位置的bed文件,表示基因注释的bed12文件。
表示基因注释时,gtf/gff和bed文件的区别
1)gtf/gff文件一行表示一个exon/CDS等子区域,多行联合表示一个gene;bed文件一行表示一个gene;
2)gtf文件中碱基位置定位方式是1-based(即起始的碱基记为1),而bed中碱基定位方式是0-based(即起始的碱基记为0)。
bed文件每一行对应信息
必须包含的3列信息:
1)chrom:染色体名字 (e.g.chr3, chrY, chr2_random或者scaffold10671)。
2)chromStart:基因在染色体或scaffold上的起始位置(0-based)。
3)chromEnd:基因在染色体或scaffold上的终止位置 (前闭后开)。
可选的9列信息:
4)name:bed文件的行名。
5)score:本条基因在注释数据集文件中的评分(0-1000),在Genome Browser中会根据不同区段的评分显示对应的阴影强度(评分越高灰度越高)。
6)strand:链的方向+、-或. (.表示不确定链的方向)
7)thickStart:CDS区(编码区)的起始位置,即起始密码子的位置。
8)thickEnd:The endingposition at which the feature is drawn thickly (for example the stop codon ingene displays).
9)itemRgb:RGB颜色值(如:255,0,0),方便在GenomeBrowser中查看。
10)blockCount:bed行中外显子的数目。
11)blockSizes:逗号分割的列,数目与blockCount值对应,每个数表示对应外显子的碱基数。
12)blockStarts:逗号分割的列,数目与blockCount值对应,每个数表示对应外显子的起始位置(数值是相对ChromStart计算的)。
bedGraph文件
Genome Browser bedGraph Track Format
BedGraph格式文件,它是BED文件的扩展,是4列的BED格式,但是需要添加UCSC的Genome Browser工具里面显示的属性,一般就定义有限的几个属性即可。
BedGraph,它的trace type和Wig文件很像,不过后面的数据和bed文件很类似,后面的四列分别表示染色体序号,起始位置,结束位置和value值。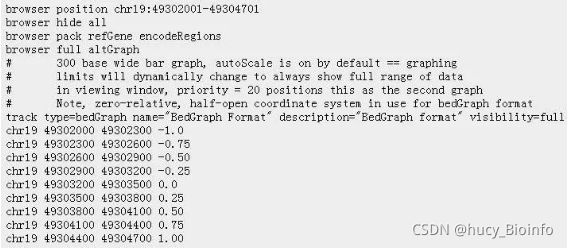
R语言和linux实现从bedgraph到bam的转化
从bedgraph到bed
以GSE139123举例,我们先下载你感兴趣的一个bedgraph文件,用R查看一下
1、GSE139123_CtrlA_peak.bedGraph放入你的工作目录
2、bedgraph<-read.table("D:/R/project_phenotypic_analysis/M6A-seq/source/GSE139123_CtrlA_peak.bedGraph")
3、View(bedgraph)
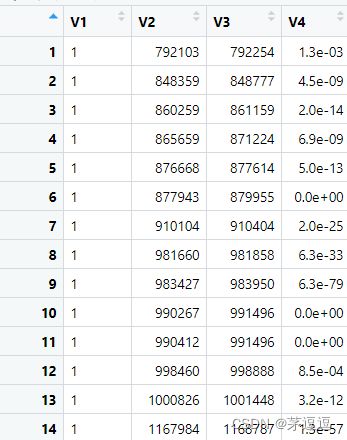
你可以观察到一共有四列,bedGraph至少有3列包含位置信息即可,第四行可要可不要。
然后直接存为Bed可以吗?答:不可以。完成如下操作才行
1、第一列改成chr1 chr10 X这样的染色体格式
2、取消行名列名
3、使用制表符\t隔开
4、删掉第一行,或者+#,这里在读取的时候把本来就有#的行干掉了,所以已经没有第一行了没在linux处理的时候,还是有的。
bedgraph<-read.table("D:/R/project_phenotypic_analysis/M6A-seq/source/GSE139123_CtrlA_peak.bedGraph")
bedgraph$V1<- str_c("chr", bedgraph$V1)
write.table (bedgraph,"c1.bed",col.names = F,row.names = F,sep = "\t")genome.size.txt
保存为bed文件后,你还需要一个参考文件:genome.size.txt
genome size文件是为了最后一步转化为bam文件所必须的,samtools可以很简单的建立index文件
# Build genome index file
samtools faidx Arabidopsis_thaliana.TAIR10.dna.toplevel.fa# Build the genome size file
awk {'print "Chr"$1,"\t",$2'} Arabidopsis_thaliana.TAIR10.dna.toplevel.fa.fai > Arabidopsis_genomeFile.txtbed转bam
bedtools工具提供的bedtobam命令
![]()
大功告成。