同源建模-build loop
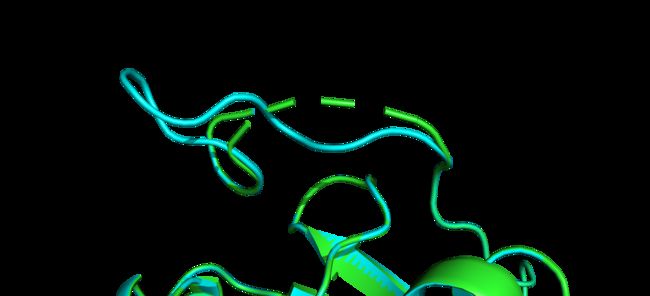
对于有残基缺失的晶体结构往往采用同源建模的方式补全,但常规的同源建模方法往往造成非缺失区域残基的挪动,有时我们并不想出现这种状况,尤其是涉及到多个截短体拼合的情况,这时就需要用到约束性同源建模的方法,只对缺失区域补全而尽可能少地改动非缺失区域。效果图如下所示:
1、准备建模序列文件
>P1;Target
sequence:Target:::::::0.00: 0.00
TCFSGDDDSGLNLGFSTSFAEYNEFDGGEKALIGFSTSFAEFDAEAGDSDEQPIFPQHKLTCFS*
前两行格式严格遵守!!!
保存文件名为Target.ali
将模板结构文件存储为temp.pdb文件(单链,除去水和离子)
以下是从pdb文件中提取fasta序列的脚本:
from Bio import PDB
from Bio.PDB import PDBParser, PDBIO
from Bio.PDB.Polypeptide import PPBuilder
from Bio.Seq import Seq
from Bio.SeqRecord import SeqRecord
import sys
def extract_fasta_from_pdb(pdb_file, type):
parser = PDBParser(QUIET=True)
if type == "pdb":
structure = parser.get_structure('PDB', pdb_file)
if type == "pdb.gz":
with gzip.open(pdb_file, 'rb') as f_in:
with open('temp.pdb', 'wb') as f_out:
f_out.write(f_in.read())
structure = parser.get_structure('PDB', 'temp.pdb')
ppb = PPBuilder()
fasta_records = []
for model in structure:
for chain in model:
polypeptides = ppb.build_peptides(chain)
for polypeptide in polypeptides:
sequence = str(polypeptide.get_sequence())
record = SeqRecord(Seq(sequence), id=chain.id)
fasta_records.append(record)
try:
os.remove('temp.pdb')
except:
pass
return fasta_records
def main():
file = open("out.fasta", "w")
sys.stdout = file
print(">temp")
pdb_file = "./temp.pdb"
s = extract_fasta_from_pdb(pdb_file, "pdb")
for record in s:
print(record.seq)
file.close()
if __name__ == "__main__":
main()
2、与模板蛋白结构进行序列比对
准备align2d.py脚本文件:
from modeller import *
env = Environ()
aln = Alignment(env)
# temp是模板结构名称,C指的是C链
mdl = Model(env, file='temp', model_segment=('FIRST:C','LAST:C'))
aln.append_model(mdl, align_codes='temp', atom_files='temp.pdb')
aln.append(file='Target.ali', align_codes='Target')
aln.align2d(max_gap_length=50)
aln.write(file='Target-temp.ali', alignment_format='PIR')
aln.write(file='Target-temp.pap', alignment_format='PAP')
执行后,当前路径下生成两个文件Target-temp.ali,Target-temp.pap。
3、 建模
准备loop_modeling.py脚本文件:
from modeller import *
from modeller.automodel import *
log.verbose()
env = Environ()
# directories for input atom files
env.io.atom_files_directory = ['.', 'temp.pdb']
class MyModel(AutoModel):
def select_atoms(self):
#新建模蛋白链编码为A,编号从1开始,因此缺失残基编号要相应挪动
#编号最好包含首尾各一个残基
return Selection(self.residue_range('5:A', '13:A'))
a = MyModel(env, alnfile = 'Target-temp.ali',
knowns = 'temp', sequence = 'Target')
a.starting_model= 1
a.ending_model = 1
a.make()
4、如何方便地确定新建模型中插入loop的编号
准备脚本文件renumber_pro.py:
import sys
from optparse import OptionParser
def initialize_options() :
parser = OptionParser();
parser.add_option( "-p", "--pdb", dest="pdbname", type="string", help="The input pdb that should be renumbered" );
parser.add_option( "-o", "--output", dest="output", type="string", help="The output file to write the renumbered pdb to; stdout if not given", default="" );
parser.add_option( "-a", "--chainA_begin", dest="chAbegin", type="int", help="The first residue in chain A", default=1 );
parser.add_option( "-b", "--chainB_begin", dest="chBbegin", type="int", help="The first residue in chain B", default=1 );
parser.add_option( "-c", "--chainC_begin", dest="chCbegin", type="int", help="The first residue in chain C", default=1 );
parser.add_option( "-d", "--chainD_begin", dest="chDbegin", type="int", help="The first residue in chain D", default=1 );
parser.add_option( "-e", "--chainE_begin", dest="chEbegin", type="int", help="The first residue in chain E", default=1 );
return parser
def remap_resnum_for_line( line, mapping ):
if line[0:4] != "ATOM": return line
chain = line[21];
if chain not in mapping: return line
resstring = line[22:27]
resnum, lastresstring = mapping[ chain ]
if lastresstring == "" or resstring != lastresstring :
if lastresstring != "" : resnum += 1
mapping[ chain ] = (resnum, resstring )
newresstring = str(resnum) + " "
if len(newresstring) == 2: newresstring = " " + newresstring
elif len(newresstring) == 3: newresstring = " " + newresstring
elif len(newresstring) == 4: newresstring = " " + newresstring
return line[0:22] + newresstring + line[27:]
if __name__ == "__main__":
parser = initialize_options()
(options, args) = parser.parse_args()
pdblines = open( options.pdbname ).readlines()
mapping = {}
mapping["A"] = (options.chAbegin, "");
mapping["B"] = (options.chBbegin, "");
mapping["C"] = (options.chCbegin, "");
mapping["D"] = (options.chDbegin, "");
mapping["E"] = (options.chEbegin, "");
newlines = []
for line in pdblines:
newlines.append( remap_resnum_for_line( line, mapping ))
if options.output == "" :
for line in newlines :
print(line, end=' ')
else :
open( options.output, "w").writelines(newlines)
执行:
python renumber_pro.py -p temp.pdb -o output.pdb
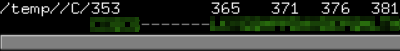
用pymol打开原模板文件发现原序列在357-365之间的残基缺失
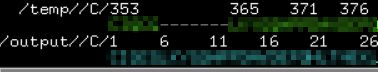
对应到重编号的文件中是5-13
因此对应上述脚本文件loop_modeling.py中对下方内容进行调整:
return Selection(self.residue_range('5:A', '13:A'))