MCScanX是另外一个检测基因共线性和进化分析的常用工具之一,2012发表至今引用数已经有好几百了,作者之一的唐海宝老师是国内植物基因组学生信分析、软件开发领域的大拿,在学习使用MCScanx之前推荐先看看他08年介绍gene synteny和collinearity概念的science文章以及MCScanX软件算法文章。
Tang H, Bowers J E, Wang X, et al. Synteny and Collinearity in Plant Genomes[J]. Science, 2008, 320(5875):486-488.
Wang Y, Tang H, DeBarry JD, Tan X, Li J, Wang X, Lee TH, Jin H, Marler B, Guo H, Kissinger JC, Paterson AH. (2012) MCScanX: a toolkit for detection and evolutionary analysis of gene synteny andcollinearity. Nucleic Acids Res, 40(7): e49.
MCScanX是MCScan的升级版本:
1)用法更简单,只需要blastp m8格式的比对文件和经过简单处理的gff作为输入文件即可;
2)参数更多元,可以设置gap的阈值;
3)输出文件中有html网页版的展示,可以看到第一列是duplicaiton depth,tandem genes用红色标出;
4)优化了算法,缓和了物种间不同gene densities的影响
一、下载和安装
从下面链接中下载:https://github.com/wyp1125/MCScanX
unzip MCscanX.zip
cd MCScanX
make
MCScanX、MCScanX_h、duplicate_gene_classifier这三个是核心程序,downstream_analyses中包含12个下游分析程序,可以画图构树,还是很方便很强大的。
make报错:
g++ struct.cc mcscan.cc read_data.cc out_utils.cc dagchainer.cc msa.cc permutation.cc -o MCScanX
msa.cc: In function ‘void msa_main(const char*)’:
msa.cc:289:22: error: ‘chdir’ was not declared in this scope
if (chdir(html_fn)<0)
^
make: *** [mcscanx] Error 1
这个错误的原因是,MCScanX 不支持64位系统。如果要在 64位上运行,需要修改下源代码。需要给MCScanX 目录下的 msa.h, dissect_multiple_alignment.h, and detect_collinear_tandem_arrays.h 这三个文件内容的最前面添加(#include
正确的做法是用 vi 打开文件,分别在三个文件( msa.h, dissect_multiple_alignment.h, and detect_collinear_tandem_arrays.h)的最前面添加:
如果还报错,检查安装是的用户,不要是root用户。不要切换到root安装,普通用户sudo安装就可以.
二、常规运行
1. 准备 gff 文件
MCscanX要求的gff文件和标准的gff文件不一样,它只有四列, 其中"sp#"的sp意味着你要用2个字母代表物种(多个字母好像也不影响结果),#则表示是哪条染色体。而"gene"则要是你蛋白序列的基因名。
可以用awk得到,第一列是物种名和染色体编号,第二列是基因号,第三列是起始位置,第四列是终止位置(用 tab 分割)

2. blastp(protein-protein BLAST)比对
我们用data中的at和vv作为例子来测试。
注:这里是找at和vv两个基因组组内和组间的共线性,因为想同时知道物种内和物种间的共线性,所以在blast之前把at和vv的基因组fasta
cat到一起,既做database,又做query,如果只想知道组间的共线性,那么就任取一个基因组为database,另一个做query。
# 合并
cat at.fa vv.fa >all.fasta
# 建库
makeblastdb -in all.fa -dbtype prot -parse_seqids -outall (-logfile allpep.log -title all)
# 蛋白比对
blastp -query all.fa -db all -out at_vv.blast -evalue 1e-10 -num_threads 16 -outfmt 6 -num_alignments 5
小tips:
- blast这一步是限速步骤,可以把all.fasta文件cut成多份,同时并行跑节省时间;
- 亲自验证该软件最多只能做5个物种的共线性。。。不管输入再多物种结果只有五个!!
得到的 at_vv.blast 文件,格式如下:
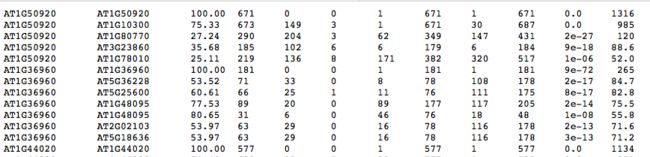
3. 运行MCScanX
输入文件只有两个,一个是blast得到的at_vv.blast文件,一个是变相的gff文件at_vv.gff。注:两个文件必须前缀相同且在同一个文件夹内
MCScanX at_vv
其他参数设置

运行成功后得到at_vv.html,at_vv.collinearity,at_vv.tandem输出文件。
at_vv.collinearity 里记录了共线性信息,可以看到collinear gene的数目和占比以及具体的比对信息
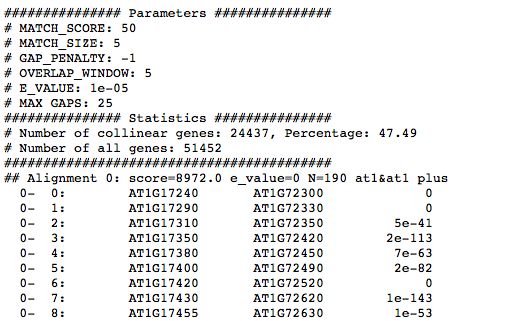
三、下游分析及可视化
常用的下游分析有:
duplicate_gene_classifier XX
- 0:singleton(非重复基因)
- 1:dispersed(不是2,3,4的其它重复)
- 2:proximal(染色体附近的重复,但是不相邻)
- 3:tandem(串联重复)
- 4:WGD/segmental(在共线性区域的共线性基因))
dissect_multiple_alignment -g XX.gff -cXX.collinearity -o XX.dis
group_collinear_genes.pl -i XX.collinearity-o XX.collinear.groups
可视化:软件自带的分析包不能调颜色,所以推荐用python版的MCscan(JCVI 包)。
这里直接使用下游dot_plotter, dual_synteny_plotter, circle_plotter和bar_plotter4个java包更便捷
gff和collinearity是上一步的输出,还需要编辑一个control文件,设置需要展示的染色体信息(和gff的第一列一致)
1. dot_plotter
dot.ctl control文件信息如下图所示:
java dot_plotter -g at_vv.gff -s at_vv.collinearity -c dot.ctl -o dot.PNG
2. dual_synteny_plotter
java dual_synteny_plotter -g at_vv.gff -s at_vv.collinearity -c dual_synteny.ctl -o dual_synteny.PNG
3. circle_plotter
java circle_plotter -g at_vv.gff -s at_vv.collinearity -c circle.ctl -o circle.PNG
这个图我更喜欢用circos去画,感觉更专业一点。
4. bar_plotter
java bar_plotter -g at_vv.gff -s at_vv.collinearity -c bar.ctl -o bar.PNG
其它分析
当然还有很多其它的分析包例如:duplicate_gene_classifier,detect_collinear_tandem_arrays,dissect_multiple_alignments对结果做进一步分析,借助其他相关信。
还可以做origin_enrichment_analysis,family_tree_plotter,add_ka_and_ks_to_collinearity等。
perl ../../downstream_analyses/add_kaks_to_synteny.pl -i at.collinearity -d at.cds -o at.collinearity.kaks
https://github.com/wyp1125/MCScanX
http://events.jianshu.io/p/519061e5d515
https://www.jianshu.com/p/d671a5b75216