手把手教你做单细胞测序数据分析(五)——细胞类型注释
前面的课程中基本已带领大家将单细胞测序预处理部分打通了,这里给大家介绍的是单细胞测序中最让人头疼的细胞类型注释。第一个代码块中没有注释,看不懂的同学去看一下我们的第三讲 单样本分析,测试数据也与那里的相同。
视频教程:
手把手教你做单细胞测序数据分析(五)——细胞类型注释
(B站同步播出,先看一遍视频再跟着代码一起操作,建议每个视频至少看三遍)
准备工作
先进行预处理,作到细胞注释前的步骤
if(T){rm(list = ls())
if (!require("Seurat"))install.packages("Seurat")
if (!require("BiocManager", quietly = TRUE))install.packages("BiocManager")
if (!require("multtest", quietly = TRUE))install.packages("multtest")
if (!require("dplyr", quietly = TRUE))install.packages("dplyr")
download.file('https://cf.10xgenomics.com/samples/cell/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz','pbmc3k_filtered_gene_bc_matrices.tar.gz')
library(R.utils)
gunzip('pbmc3k_filtered_gene_bc_matrices.tar.gz')
untar('pbmc3k_filtered_gene_bc_matrices.tar')
pbmc.data <- Read10X('filtered_gene_bc_matrices/hg19/')
pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200)
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)
pbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize", scale.factor = 10000)
pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)
top10 <- head(VariableFeatures(pbmc), 10)
pbmc <- ScaleData(pbmc, features = rownames(pbmc))
pbmc <- ScaleData(pbmc, vars.to.regress = "percent.mt")
pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc))
print(pbmc[["pca"]], dims = 1:5, nfeatures = 5)
VizDimLoadings(pbmc, dims = 1:2, reduction = "pca")
DimHeatmap(pbmc, dims = 1, cells = 500, balanced = TRUE)
DimHeatmap(pbmc, dims = 1:15, cells = 500, balanced = TRUE)
pbmc <- JackStraw(pbmc, num.replicate = 100)
pbmc <- ScoreJackStraw(pbmc, dims = 1:20)
JackStrawPlot(pbmc, dims = 1:15)
pbmc <- FindNeighbors(pbmc, dims = 1:10)
pbmc <- FindClusters(pbmc, resolution = 0.5)
head(Idents(pbmc), 5)
pbmc <- RunUMAP(pbmc, dims = 1:10)
pbmc <- RunTSNE(pbmc, dims = 1:10)
DimPlot(pbmc, reduction = "umap", label = TRUE)
pbmc.markers <- FindAllMarkers(pbmc, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)
library(dplyr)
pbmc.markers %>% group_by(cluster) %>% top_n(n = 2, wt = avg_log2FC)
}
## Loading required package: Seurat
## Warning: package 'Seurat' was built under R version 4.0.5
## Attaching SeuratObject
## Warning: package 'BiocManager' was built under R version 4.0.5
## Bioconductor version '3.12' is out-of-date; the current release version '3.13'
## is available with R version '4.1'; see https://bioconductor.org/install
## Warning: package 'BiocGenerics' was built under R version 4.0.5
##
## Attaching package: 'BiocGenerics'
## The following objects are masked from 'package:parallel':
##
## clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
## clusterExport, clusterMap, parApply, parCapply, parLapply,
## parLapplyLB, parRapply, parSapply, parSapplyLB
## The following objects are masked from 'package:stats':
##
## IQR, mad, sd, var, xtabs
## The following objects are masked from 'package:base':
##
## anyDuplicated, append, as.data.frame, basename, cbind, colnames,
## dirname, do.call, duplicated, eval, evalq, Filter, Find, get, grep,
## grepl, intersect, is.unsorted, lapply, Map, mapply, match, mget,
## order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
## rbind, Reduce, rownames, sapply, setdiff, sort, table, tapply,
## union, unique, unsplit, which.max, which.min
## Welcome to Bioconductor
##
## Vignettes contain introductory material; view with
## 'browseVignettes()'. To cite Bioconductor, see
## 'citation("Biobase")', and for packages 'citation("pkgname")'.
## Warning: package 'dplyr' was built under R version 4.0.5
##
## Attaching package: 'dplyr'
## The following object is masked from 'package:Biobase':
##
## combine
## The following objects are masked from 'package:BiocGenerics':
##
## combine, intersect, setdiff, union
## The following objects are masked from 'package:stats':
##
## filter, lag
## The following objects are masked from 'package:base':
##
## intersect, setdiff, setequal, union
## Loading required package: R.oo
## Loading required package: R.methodsS3
## R.methodsS3 v1.8.1 (2020-08-26 16:20:06 UTC) successfully loaded. See ?R.methodsS3 for help.
## R.oo v1.24.0 (2020-08-26 16:11:58 UTC) successfully loaded. See ?R.oo for help.
##
## Attaching package: 'R.oo'
## The following object is masked from 'package:R.methodsS3':
##
## throw
## The following objects are masked from 'package:methods':
##
## getClasses, getMethods
## The following objects are masked from 'package:base':
##
## attach, detach, load, save
## R.utils v2.10.1 (2020-08-26 22:50:31 UTC) successfully loaded. See ?R.utils for help.
##
## Attaching package: 'R.utils'
## The following object is masked from 'package:utils':
##
## timestamp
## The following objects are masked from 'package:base':
##
## cat, commandArgs, getOption, inherits, isOpen, nullfile, parse,
## warnings
## Warning: Feature names cannot have underscores ('_'), replacing with dashes
## ('-')
## Centering and scaling data matrix
## Regressing out percent.mt
## Centering and scaling data matrix
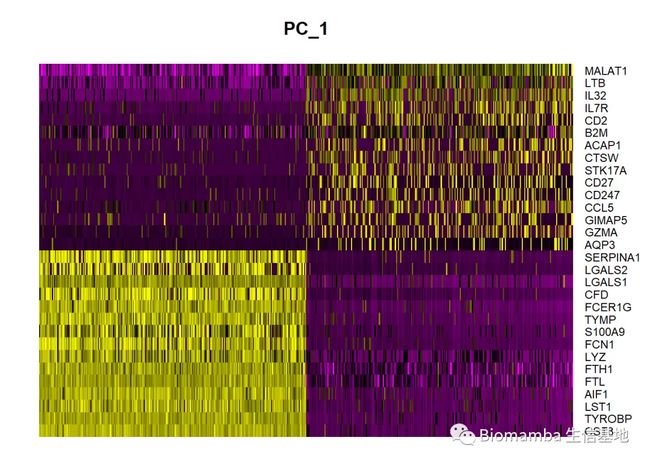
## PC_ 1
## Positive: CST3, TYROBP, LST1, AIF1, FTL, FTH1, LYZ, FCN1, S100A9, TYMP
## FCER1G, CFD, LGALS1, LGALS2, SERPINA1, S100A8, CTSS, IFITM3, SPI1, CFP
## PSAP, IFI30, COTL1, SAT1, S100A11, NPC2, GRN, LGALS3, GSTP1, PYCARD
## Negative: MALAT1, LTB, IL32, IL7R, CD2, B2M, ACAP1, CTSW, STK17A, CD27
## CD247, CCL5, GIMAP5, GZMA, AQP3, CST7, TRAF3IP3, SELL, GZMK, HOPX
## MAL, MYC, ITM2A, ETS1, LYAR, GIMAP7, KLRG1, NKG7, ZAP70, BEX2
## PC_ 2
## Positive: CD79A, MS4A1, TCL1A, HLA-DQA1, HLA-DQB1, HLA-DRA, LINC00926, CD79B, HLA-DRB1, CD74
## HLA-DMA, HLA-DPB1, HLA-DQA2, CD37, HLA-DRB5, HLA-DMB, HLA-DPA1, FCRLA, HVCN1, LTB
## BLNK, P2RX5, IGLL5, IRF8, SWAP70, ARHGAP24, FCGR2B, SMIM14, PPP1R14A, C16orf74
## Negative: NKG7, PRF1, CST7, GZMA, GZMB, FGFBP2, CTSW, GNLY, B2M, SPON2
## CCL4, GZMH, FCGR3A, CCL5, CD247, XCL2, CLIC3, AKR1C3, SRGN, HOPX
## TTC38, CTSC, APMAP, S100A4, IGFBP7, ANXA1, ID2, IL32, XCL1, RHOC
## PC_ 3
## Positive: HLA-DQA1, CD79A, CD79B, HLA-DQB1, HLA-DPA1, HLA-DPB1, CD74, MS4A1, HLA-DRB1, HLA-DRA
## HLA-DRB5, HLA-DQA2, TCL1A, LINC00926, HLA-DMB, HLA-DMA, CD37, HVCN1, FCRLA, IRF8
## PLAC8, BLNK, MALAT1, SMIM14, PLD4, IGLL5, SWAP70, P2RX5, LAT2, FCGR3A
## Negative: PPBP, PF4, SDPR, SPARC, GNG11, NRGN, GP9, RGS18, TUBB1, CLU
## HIST1H2AC, AP001189.4, ITGA2B, CD9, TMEM40, PTCRA, CA2, ACRBP, MMD, TREML1
## NGFRAP1, F13A1, SEPT5, RUFY1, TSC22D1, CMTM5, MPP1, MYL9, RP11-367G6.3, GP1BA
## PC_ 4
## Positive: HLA-DQA1, CD79B, CD79A, MS4A1, HLA-DQB1, CD74, HLA-DPB1, HIST1H2AC, HLA-DPA1, HLA-DRB1
## TCL1A, PF4, HLA-DQA2, SDPR, HLA-DRA, LINC00926, PPBP, GNG11, HLA-DRB5, SPARC
## GP9, PTCRA, CA2, AP001189.4, CD9, NRGN, RGS18, GZMB, CLU, TUBB1
## Negative: VIM, IL7R, S100A6, S100A8, IL32, S100A4, GIMAP7, S100A10, S100A9, MAL
## AQP3, CD14, CD2, LGALS2, FYB, GIMAP4, ANXA1, RBP7, CD27, FCN1
## LYZ, S100A12, MS4A6A, GIMAP5, S100A11, FOLR3, TRABD2A, AIF1, IL8, TMSB4X
## PC_ 5
## Positive: GZMB, FGFBP2, S100A8, NKG7, GNLY, CCL4, PRF1, CST7, SPON2, GZMA
## GZMH, LGALS2, S100A9, CCL3, XCL2, CD14, CLIC3, CTSW, MS4A6A, GSTP1
## S100A12, RBP7, IGFBP7, FOLR3, AKR1C3, TYROBP, CCL5, TTC38, XCL1, APMAP
## Negative: LTB, IL7R, CKB, MS4A7, RP11-290F20.3, AQP3, SIGLEC10, VIM, CYTIP, HMOX1
## LILRB2, PTGES3, HN1, CD2, FAM110A, CD27, ANXA5, CTD-2006K23.1, MAL, VMO1
## CORO1B, TUBA1B, LILRA3, GDI2, TRADD, ATP1A1, IL32, ABRACL, CCDC109B, PPA1
## PC_ 1
## Positive: CST3, TYROBP, LST1, AIF1, FTL
## Negative: MALAT1, LTB, IL32, IL7R, CD2
## PC_ 2
## Positive: CD79A, MS4A1, TCL1A, HLA-DQA1, HLA-DQB1
## Negative: NKG7, PRF1, CST7, GZMA, GZMB
## PC_ 3
## Positive: HLA-DQA1, CD79A, CD79B, HLA-DQB1, HLA-DPA1
## Negative: PPBP, PF4, SDPR, SPARC, GNG11
## PC_ 4
## Positive: HLA-DQA1, CD79B, CD79A, MS4A1, HLA-DQB1
## Negative: VIM, IL7R, S100A6, S100A8, IL32
## PC_ 5
## Positive: GZMB, FGFBP2, S100A8, NKG7, GNLY
## Negative: LTB, IL7R, CKB, MS4A7, RP11-290F20.3
## Computing nearest neighbor graph
## Computing SNN
## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
##
## Number of nodes: 2638
## Number of edges: 95893
##
## Running Louvain algorithm...
## Maximum modularity in 10 random starts: 0.8735
## Number of communities: 9
## Elapsed time: 0 seconds
## Warning: The default method for RunUMAP has changed from calling Python UMAP via reticulate to the R-native UWOT using the cosine metric
## To use Python UMAP via reticulate, set umap.method to 'umap-learn' and metric to 'correlation'
## This message will be shown once per session
## 09:15:44 UMAP embedding parameters a = 0.9922 b = 1.112
## 09:15:44 Read 2638 rows and found 10 numeric columns
## 09:15:44 Using Annoy for neighbor search, n_neighbors = 30
## 09:15:44 Building Annoy index with metric = cosine, n_trees = 50
## 0% 10 20 30 40 50 60 70 80 90 100%
## [----|----|----|----|----|----|----|----|----|----|
## **************************************************|
## 09:15:44 Writing NN index file to temp file C:\Users\53513\AppData\Local\Temp\Rtmpo9Xcsm\file56685416913
## 09:15:44 Searching Annoy index using 1 thread, search_k = 3000
## 09:15:45 Annoy recall = 100%
## 09:15:45 Commencing smooth kNN distance calibration using 1 thread
## 09:15:46 Initializing from normalized Laplacian + noise
## 09:15:46 Commencing optimization for 500 epochs, with 106338 positive edges
## 09:15:52 Optimization finished
## Calculating cluster 0
## Calculating cluster 1
## Calculating cluster 2
## Calculating cluster 3
## Calculating cluster 4
## Calculating cluster 5
## Calculating cluster 6
## Calculating cluster 7
## Calculating cluster 8

## Registered S3 method overwritten by 'cli':
## method from
## print.boxx spatstat.geom
## # A tibble: 18 x 7
## # Groups: cluster [9]
## p_val avg_log2FC pct.1 pct.2 p_val_adj cluster gene
##
## 1 1.88e-117 1.08 0.913 0.588 2.57e-113 0 LDHB
## 2 5.01e- 85 1.34 0.437 0.108 6.88e- 81 0 CCR7
## 3 0 5.57 0.996 0.215 0 1 S100A9
## 4 0 5.48 0.975 0.121 0 1 S100A8
## 5 1.93e- 80 1.27 0.981 0.65 2.65e- 76 2 LTB
## 6 2.91e- 58 1.27 0.667 0.251 3.98e- 54 2 CD2
## 7 0 4.31 0.939 0.042 0 3 CD79A
## 8 1.06e-269 3.59 0.623 0.022 1.45e-265 3 TCL1A
## 9 3.60e-221 3.21 0.984 0.226 4.93e-217 4 CCL5
## 10 4.27e-176 3.01 0.573 0.05 5.85e-172 4 GZMK
## 11 3.51e-184 3.31 0.975 0.134 4.82e-180 5 FCGR3A
## 12 2.03e-125 3.09 1 0.315 2.78e-121 5 LST1
## 13 3.17e-267 4.83 0.961 0.068 4.35e-263 6 GZMB
## 14 1.04e-189 5.28 0.961 0.132 1.43e-185 6 GNLY
## 15 1.48e-220 3.87 0.812 0.011 2.03e-216 7 FCER1A
## 16 1.67e- 21 2.87 1 0.513 2.28e- 17 7 HLA-DPB1
## 17 7.73e-200 7.24 1 0.01 1.06e-195 8 PF4
## 18 3.68e-110 8.58 1 0.024 5.05e-106 8 PPBP
方法一:查数据库
library(dplyr)
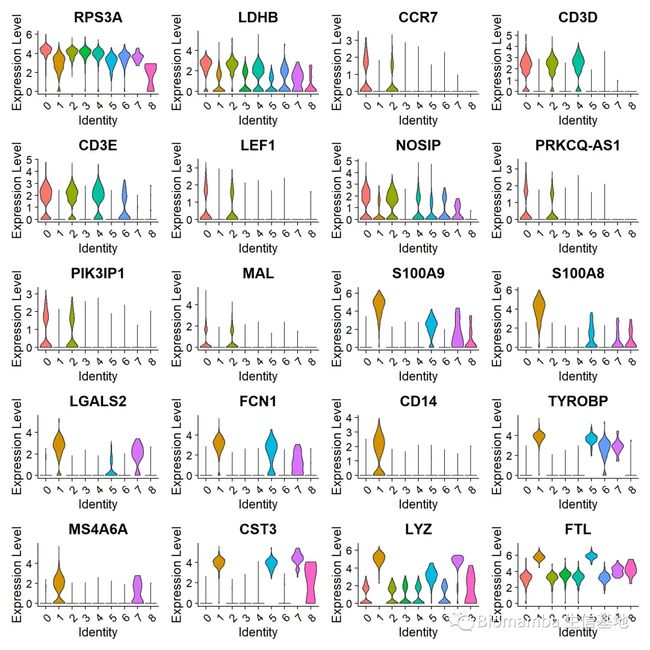
top10 <- pbmc.markers %>% group_by(cluster) %>% top_n(n = 10, wt = avg_log2FC)
DoHeatmap(pbmc, features = top10$gene) + NoLegend()#展示前10个标记基因的热图
## Warning in DoHeatmap(pbmc, features = top10$gene): The following features were
## omitted as they were not found in the scale.data slot for the RNA assay: CD8A,
## VPREB3, CD40LG, PIK3IP1, PRKCQ-AS1, NOSIP, LEF1, CD3E, CD3D, CCR7, LDHB, RPS3A
VlnPlot(pbmc, features = top10$gene[1:20],pt.size=0)
DimPlot(pbmc,label = T)
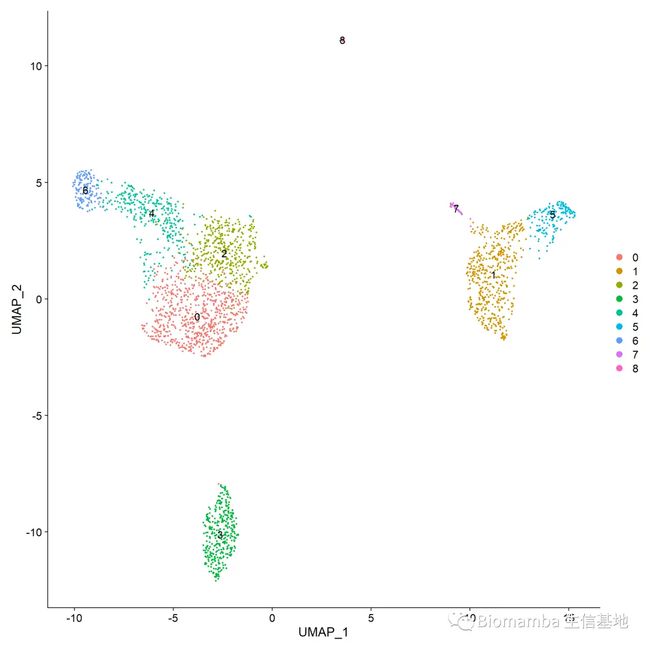
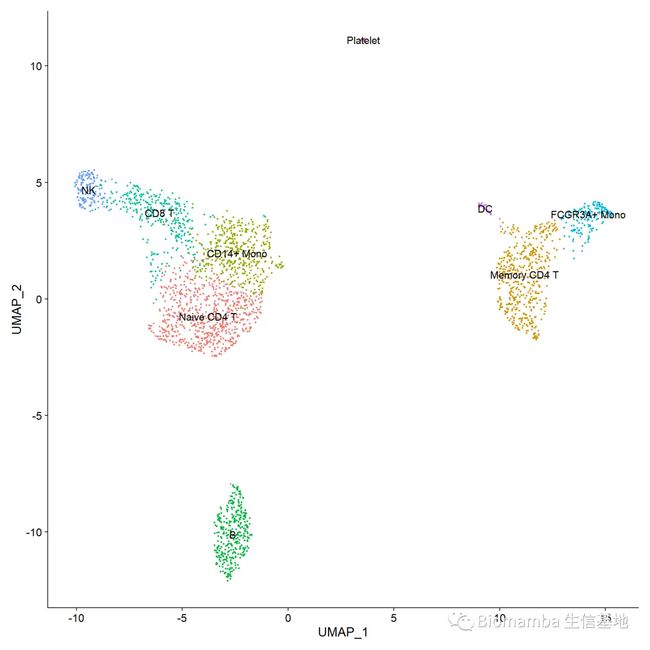
#通过标记基因及文献,可以人工确定各分类群的细胞类型,则可以如下手动添加细胞群名称
bfreaname.pbmc <- pbmc
new.cluster.ids <- c("Naive CD4 T", "Memory CD4 T", "CD14+ Mono", "B", "CD8 T", "FCGR3A+ Mono",
"NK", "DC", "Platelet")
#帮助单细胞测序进行注释的数据库:
#http://mp.weixin.qq.com/s?__biz=MzI5MTcwNjA4NQ==&mid=2247502903&idx=2&sn=fd21e6e111f57a4a2b6c987e391068fd&chksm=ec0e09bddb7980abf038f62d03d3beea6249753c8fba69b69f399de9854fc208ca863ca5bc23&mpshare=1&scene=24&srcid=1110SJhxDL8hmNB5BThrgOS9&sharer_sharetime=1604979334616&sharer_shareid=853c5fb0f1636baa0a65973e8b5db684#rd
#cellmarker: http://biocc.hrbmu.edu.cn/CellMarker/index.jsp
names(new.cluster.ids) <- levels(pbmc)
pbmc <- RenameIdents(pbmc, new.cluster.ids)
DimPlot(pbmc, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend()
方法二:通过singleR
这一方法很鸡肋,主要是由于很难找到合适的参考数据,但是对于免疫细胞的注释还是有可观效果的
if(!require(SingleR))BiocManager::install(SingleR)
## Loading required package: SingleR
## Loading required package: SummarizedExperiment
## Loading required package: MatrixGenerics
## Loading required package: matrixStats
## Warning: package 'matrixStats' was built under R version 4.0.5
##
## Attaching package: 'matrixStats'
## The following object is masked from 'package:dplyr':
##
## count
## The following objects are masked from 'package:Biobase':
##
## anyMissing, rowMedians
##
## Attaching package: 'MatrixGenerics'
## The following objects are masked from 'package:matrixStats':
##
## colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse,
## colCounts, colCummaxs, colCummins, colCumprods, colCumsums,
## colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs,
## colMads, colMaxs, colMeans2, colMedians, colMins, colOrderStats,
## colProds, colQuantiles, colRanges, colRanks, colSdDiffs, colSds,
## colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads,
## colWeightedMeans, colWeightedMedians, colWeightedSds,
## colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet,
## rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods,
## rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps,
## rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins,
## rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks,
## rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars,
## rowWeightedMads, rowWeightedMeans, rowWeightedMedians,
## rowWeightedSds, rowWeightedVars
## The following object is masked from 'package:Biobase':
##
## rowMedians
## Loading required package: GenomicRanges
## Loading required package: stats4
## Loading required package: S4Vectors
##
## Attaching package: 'S4Vectors'
## The following objects are masked from 'package:dplyr':
##
## first, rename
## The following object is masked from 'package:base':
##
## expand.grid
## Loading required package: IRanges
##
## Attaching package: 'IRanges'
## The following object is masked from 'package:R.oo':
##
## trim
## The following objects are masked from 'package:dplyr':
##
## collapse, desc, slice
## The following object is masked from 'package:grDevices':
##
## windows
## Loading required package: GenomeInfoDb
## Warning: package 'GenomeInfoDb' was built under R version 4.0.5
##
## Attaching package: 'SummarizedExperiment'
## The following object is masked from 'package:SeuratObject':
##
## Assays
## The following object is masked from 'package:Seurat':
##
## Assays
# remove.packages('matrixStats')#移除后可能需要重新打开
if(!require(matrixStats))BiocManager::install('matrixStats')
# remove.packages(c('dplyr','ellipsis'))
# install.packages(c('dplyr','ellipsis'))
# remove.packages(c('vctrs'))
# install.packages(c('vctrs'))
if(!require(celldex))BiocManager::install('celldex')#有一些版本冲突,需要重新安装一些包。
## Loading required package: celldex
##
## Attaching package: 'celldex'
## The following objects are masked from 'package:SingleR':
##
## BlueprintEncodeData, DatabaseImmuneCellExpressionData,
## HumanPrimaryCellAtlasData, ImmGenData, MonacoImmuneData,
## MouseRNAseqData, NovershternHematopoieticData
###内置数据集:
# cg=BlueprintEncodeData()
# cg=DatabaseImmuneCellExpressionData()
# cg=NovershternHematopoieticData()
# cg=MonacoImmuneData()
# cg=ImmGenData()
# cg=MouseRNAseqData()
# cg=HumanPrimaryCellAtlasData()
cg<-ImmGenData()#选取我们要使用的免疫细胞参考数据集
## snapshotDate(): 2020-10-27
## see ?celldex and browseVignettes('celldex') for documentation
## Could not check id: EH3494 for updates.
## Using previously cached version.
## loading from cache
## Could not check id: EH3494 for updates.
## Using previously cached version.
## see ?celldex and browseVignettes('celldex') for documentation
## Could not check id: EH3495 for updates.
## Using previously cached version.
## loading from cache
## Could not check id: EH3495 for updates.
## Using previously cached version.
assay_for_SingleR <- GetAssayData(bfreaname.pbmc, slot="data")#取出样本中的表达序列
predictions <- SingleR(test=assay_for_SingleR,
ref=cg, labels=cg$label.main)
#以kidney中提取的阵列为输入数据,以小鼠的阵列作为参考,predict细胞类型
table(predictions$labels)#看看都注释到了哪些细胞
##
## B cells Endothelial cells Eosinophils Mast cells
## 2555 19 18 25
## NK cells Stromal cells
## 11 10
cellType=data.frame([email protected]$seurat_clusters,
predict=predictions$labels)#得到seurat中编号与预测标签之间的关系
sort(table(cellType[,1]))
##
## 8 7 6 5 4 3 2 1 0
## 14 32 154 162 316 342 429 480 709
table(cellType[,1:2])#访问celltyple的2~3列
## predict
## seurat B cells Endothelial cells Eosinophils Mast cells NK cells Stromal cells
## 0 696 5 4 0 3 1
## 1 458 2 6 12 2 0
## 2 415 4 6 0 2 2
## 3 342 0 0 0 0 0
## 4 311 4 0 0 1 0
## 5 137 3 1 13 1 7
## 6 151 1 1 0 1 0
## 7 31 0 0 0 1 0
## 8 14 0 0 0 0 0
#可以看出 singleR如果没有合适的数据集,得到的结果有多拉跨了吧
#这里就没必要再往下重命名了
你有合适的参考数据集时可以用方法三、方法四进行注释
方法三:自定义singleR注释
#我们走个极端,拿它自己作为自己的参考数据集,看看注释的准不准
##########利用singleR构建自己的数据作为参考数据集########
library(SingleR)
library(Seurat)
library(ggplot2)
## Warning: package 'ggplot2' was built under R version 4.0.5
if(!require(textshape))install.packages('textshape')
## Loading required package: textshape
## Warning: package 'textshape' was built under R version 4.0.5
##
## Attaching package: 'textshape'
## The following object is masked from 'package:dplyr':
##
## combine
## The following object is masked from 'package:Biobase':
##
## combine
## The following object is masked from 'package:BiocGenerics':
##
## combine
if(!require(scater))BiocManager::install('scater')
## Loading required package: scater
## Warning: package 'scater' was built under R version 4.0.4
## Loading required package: SingleCellExperiment
if(!require(SingleCellExperiment))BiocManager::install('SingleCellExperiment')
library(dplyr)
# 读入scRNA数据 -------
myref <- pbmc##这里为了检测,我们将参考数据集与目标数据集用同一个数据进行测试
myref$celltype <- Idents(myref)
table(Idents(myref))
##
## Naive CD4 T Memory CD4 T CD14+ Mono B CD8 T FCGR3A+ Mono
## 709 480 429 342 316 162
## NK DC Platelet
## 154 32 14
# 读入参考数据集 -------
Refassay <- log1p(AverageExpression(myref, verbose = FALSE)$RNA)#求
#Ref <- textshape::column_to_rownames(Ref, loc = 1)#另一种得到参考矩阵的办法
head(Refassay)#看看表达矩阵长啥样
## Naive CD4 T Memory CD4 T CD14+ Mono B CD8 T
## AL627309.1 0.006006857 0.04740195 0.006651185 0.00000000 0.01746659
## AP006222.2 0.000000000 0.01082590 0.009196592 0.00000000 0.01016628
## RP11-206L10.2 0.007300235 0.00000000 0.000000000 0.02055829 0.00000000
## RP11-206L10.9 0.000000000 0.01044641 0.000000000 0.00000000 0.00000000
## LINC00115 0.014803993 0.03685000 0.033640559 0.03836728 0.01657028
## NOC2L 0.410974333 0.24101294 0.312227749 0.46371195 0.39676059
## FCGR3A+ Mono NK DC Platelet
## AL627309.1 0.00000000 0.00000000 0.0000000 0
## AP006222.2 0.00000000 0.00000000 0.0000000 0
## RP11-206L10.2 0.00000000 0.00000000 0.0812375 0
## RP11-206L10.9 0.01192865 0.00000000 0.0000000 0
## LINC00115 0.01458683 0.05726061 0.0000000 0
## NOC2L 0.40564359 0.53378022 0.2841343 0
ref_sce <- SingleCellExperiment::SingleCellExperiment(assays=list(counts=Refassay))
#参考数据集需要构建成一个SingleCellExperiment对象
ref_sce=scater::logNormCounts(ref_sce)
logcounts(ref_sce)[1:4,1:4]
## Naive CD4 T Memory CD4 T CD14+ Mono B
## AL627309.1 0.009250892 0.06711500 0.009538416 0.00000000
## AP006222.2 0.000000000 0.01560549 0.013172144 0.00000000
## RP11-206L10.2 0.011235027 0.00000000 0.000000000 0.02967623
## RP11-206L10.9 0.000000000 0.01506130 0.000000000 0.00000000
colData(ref_sce)$Type=colnames(Refassay)
ref_sce#构建完成
## class: SingleCellExperiment
## dim: 13714 9
## metadata(0):
## assays(2): counts logcounts
## rownames(13714): AL627309.1 AP006222.2 ... PNRC2.1 SRSF10.1
## rowData names(0):
## colnames(9): Naive CD4 T Memory CD4 T ... DC Platelet
## colData names(2): sizeFactor Type
## reducedDimNames(0):
## altExpNames(0):
###提取自己的单细胞矩阵##########
testdata <- GetAssayData(bfreaname.pbmc, slot="data")
pred <- SingleR(test=testdata, ref=ref_sce,
labels=ref_sce$Type,
#clusters = [email protected]
)
table(pred$labels)
##
## B CD14+ Mono CD8 T DC FCGR3A+ Mono Memory CD4 T
## 344 537 308 31 179 464
## Naive CD4 T NK Platelet
## 601 160 14
head(pred)
## DataFrame with 6 rows and 5 columns
## scores first.labels tuning.scores
##
## AAACATACAACCAC-1 0.336675:0.424807:0.430908:... CD8 T 0.321456:0.2879031
## AAACATTGAGCTAC-1 0.494458:0.381603:0.391545:... B 0.494458:0.4012752
## AAACATTGATCAGC-1 0.373678:0.519949:0.489834:... CD14+ Mono 0.411459:0.3466608
## AAACCGTGCTTCCG-1 0.341852:0.362193:0.353068:... Memory CD4 T 0.422782:0.3899934
## AAACCGTGTATGCG-1 0.233921:0.279848:0.329152:... NK 0.384988:0.0678734
## AAACGCACTGGTAC-1 0.391590:0.458438:0.426201:... CD14+ Mono 0.335794:0.2679515
## labels pruned.labels
##
## AAACATACAACCAC-1 CD8 T CD8 T
## AAACATTGAGCTAC-1 B B
## AAACATTGATCAGC-1 CD14+ Mono CD14+ Mono
## AAACCGTGCTTCCG-1 FCGR3A+ Mono FCGR3A+ Mono
## AAACCGTGTATGCG-1 NK NK
## AAACGCACTGGTAC-1 CD14+ Mono CD14+ Mono
as.data.frame(table(pred$labels))
## Var1 Freq
## 1 B 344
## 2 CD14+ Mono 537
## 3 CD8 T 308
## 4 DC 31
## 5 FCGR3A+ Mono 179
## 6 Memory CD4 T 464
## 7 Naive CD4 T 601
## 8 NK 160
## 9 Platelet 14
#pred@listData[["scores"]] #预测评分,想看看结构的可以自己看看
#同上,我们找一下seurat中类群与注释结果直接的关系
cellType=data.frame([email protected]$seurat_clusters,
predict=pred$labels)#得到seurat中编号与预测标签之间的关系
sort(table(cellType[,1]))
##
## 8 7 6 5 4 3 2 1 0
## 14 32 154 162 316 342 429 480 709
table(cellType[,1:2])#访问celltyple的2~3列
## predict
## seurat B CD14+ Mono CD8 T DC FCGR3A+ Mono Memory CD4 T Naive CD4 T NK
## 0 2 159 13 0 0 0 535 0
## 1 0 0 0 1 17 462 0 0
## 2 0 355 10 0 0 0 64 0
## 3 341 1 0 0 0 0 0 0
## 4 1 22 282 0 0 0 2 9
## 5 0 0 0 0 162 0 0 0
## 6 0 0 3 0 0 0 0 151
## 7 0 0 0 30 0 2 0 0
## 8 0 0 0 0 0 0 0 0
## predict
## seurat Platelet
## 0 0
## 1 0
## 2 0
## 3 0
## 4 0
## 5 0
## 6 0
## 7 0
## 8 14
lalala <- as.data.frame(table(cellType[,1:2]))
finalmap <- lalala %>% group_by(seurat) %>% top_n(n = 1, wt = Freq)#找出每种seurat_cluster注释比例最高的对应类型
finalmap <-finalmap[order(finalmap$seurat),]$predict#找到seurat中0:8的对应预测细胞类型
print(finalmap)
## [1] Naive CD4 T Memory CD4 T CD14+ Mono B CD8 T
## [6] FCGR3A+ Mono NK DC Platelet
## 9 Levels: B CD14+ Mono CD8 T DC FCGR3A+ Mono Memory CD4 T Naive CD4 T ... Platelet
testname <- bfreaname.pbmc
new.cluster.ids <- as.character(finalmap)
names(new.cluster.ids) <- levels(testname)
testname <- RenameIdents(testname, new.cluster.ids)
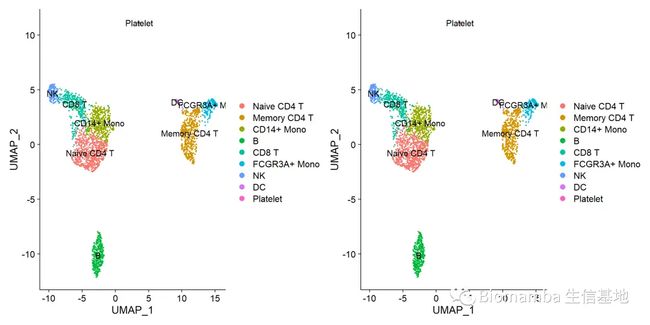
p1 <- DimPlot(pbmc,label = T)
p2 <- DimPlot(testname,label = T)#比较一下测试数据与参考数据集之间有没有偏差
p1|p2#完美,无差别注释,当然了,我们这个参考数据用的比较极端
方法四、Seurat内置的投影
######利用seurat内置的原先用于细胞整合的功能,将参考数据与待注释数据进行映射处理
library(Seurat)
pancreas.query <- bfreaname.pbmc#待注释数据
pancreas.anchors <- FindTransferAnchors(reference = pbmc, query = pancreas.query,
dims = 1:30)
## Performing PCA on the provided reference using 2000 features as input.
## Projecting cell embeddings
## Finding neighborhoods
## Finding anchors
## Found 7206 anchors
## Filtering anchors
## Retained 5268 anchors
pbmc$celltype <- Idents(pbmc)
predictions <- TransferData(anchorset = pancreas.anchors, refdata = pbmc$celltype,
dims = 1:30)
## Finding integration vectors
## Finding integration vector weights
## Predicting cell labels
pancreas.query <- AddMetaData(pancreas.query, metadata = predictions)
#把注释加回原来的数据集
pancreas.query$prediction.match <- pancreas.query$predicted.id
table(pancreas.query$prediction.match)
##
## B CD14+ Mono CD8 T DC FCGR3A+ Mono Memory CD4 T
## 340 431 294 32 158 485
## Naive CD4 T NK Platelet
## 732 153 13
Idents(pancreas.query)<- 'prediction.match'
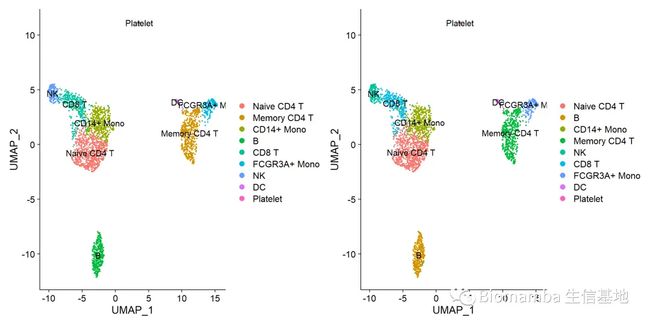
p1 <- DimPlot(pbmc,label = T)
p2 <- DimPlot(pancreas.query,label = T)#比较一下测试数据与参考数据集之间有没有偏差
p1|p2#完美,无差别注释,当然了,我们这个参考数据用的比较极端
版本问题
#### 最近发现代码运行的最大障碍是各个packages的版本冲突问题,这里列出本次分析环境中的所有信息,报错时可以参考
sessionInfo()
## R version 4.0.3 (2020-10-10)
## Platform: x86_64-w64-mingw32/x64 (64-bit)
## Running under: Windows 10 x64 (build 19042)
##
## Matrix products: default
##
## locale:
## [1] LC_COLLATE=Chinese (Simplified)_China.936
## [2] LC_CTYPE=Chinese (Simplified)_China.936
## [3] LC_MONETARY=Chinese (Simplified)_China.936
## [4] LC_NUMERIC=C
## [5] LC_TIME=Chinese (Simplified)_China.936
##
## attached base packages:
## [1] stats4 parallel stats graphics grDevices utils datasets
## [8] methods base
##
## other attached packages:
## [1] scater_1.18.6 SingleCellExperiment_1.12.0
## [3] textshape_1.7.3 ggplot2_3.3.5
## [5] celldex_1.0.0 SingleR_1.4.1
## [7] SummarizedExperiment_1.20.0 GenomicRanges_1.42.0
## [9] GenomeInfoDb_1.26.7 IRanges_2.24.1
## [11] S4Vectors_0.28.1 MatrixGenerics_1.2.1
## [13] matrixStats_0.60.1 R.utils_2.10.1
## [15] R.oo_1.24.0 R.methodsS3_1.8.1
## [17] dplyr_1.0.7 multtest_2.46.0
## [19] Biobase_2.50.0 BiocGenerics_0.36.1
## [21] BiocManager_1.30.16 SeuratObject_4.0.2
## [23] Seurat_4.0.2
##
## loaded via a namespace (and not attached):
## [1] utf8_1.2.2 reticulate_1.20
## [3] tidyselect_1.1.1 RSQLite_2.2.8
## [5] AnnotationDbi_1.52.0 htmlwidgets_1.5.4
## [7] grid_4.0.3 BiocParallel_1.24.1
## [9] Rtsne_0.15 munsell_0.5.0
## [11] codetools_0.2-16 ica_1.0-2
## [13] future_1.22.1 miniUI_0.1.1.1
## [15] withr_2.4.2 colorspace_2.0-2
## [17] highr_0.9 knitr_1.34
## [19] rstudioapi_0.13 ROCR_1.0-11
## [21] tensor_1.5 listenv_0.8.0
## [23] labeling_0.4.2 GenomeInfoDbData_1.2.4
## [25] polyclip_1.10-0 bit64_4.0.5
## [27] farver_2.1.0 parallelly_1.28.1
## [29] vctrs_0.3.8 generics_0.1.0
## [31] xfun_0.25 BiocFileCache_1.14.0
## [33] R6_2.5.1 ggbeeswarm_0.6.0
## [35] rsvd_1.0.5 bitops_1.0-7
## [37] spatstat.utils_2.2-0 cachem_1.0.6
## [39] DelayedArray_0.16.3 assertthat_0.2.1
## [41] promises_1.2.0.1 scales_1.1.1
## [43] beeswarm_0.4.0 gtable_0.3.0
## [45] beachmat_2.6.4 globals_0.14.0
## [47] goftest_1.2-2 rlang_0.4.11
## [49] splines_4.0.3 lazyeval_0.2.2
## [51] spatstat.geom_2.2-2 yaml_2.2.1
## [53] reshape2_1.4.4 abind_1.4-5
## [55] httpuv_1.6.2 tools_4.0.3
## [57] ellipsis_0.3.2 spatstat.core_2.3-0
## [59] jquerylib_0.1.4 RColorBrewer_1.1-2
## [61] ggridges_0.5.3 Rcpp_1.0.7
## [63] plyr_1.8.6 sparseMatrixStats_1.2.1
## [65] zlibbioc_1.36.0 purrr_0.3.4
## [67] RCurl_1.98-1.4 rpart_4.1-15
## [69] deldir_0.2-10 viridis_0.6.1
## [71] pbapply_1.4-3 cowplot_1.1.1
## [73] zoo_1.8-9 ggrepel_0.9.1
## [75] cluster_2.1.0 magrittr_2.0.1
## [77] data.table_1.14.0 RSpectra_0.16-0
## [79] scattermore_0.7 lmtest_0.9-38
## [81] RANN_2.6.1 fitdistrplus_1.1-5
## [83] patchwork_1.1.1 mime_0.11
## [85] evaluate_0.14 xtable_1.8-4
## [87] gridExtra_2.3 compiler_4.0.3
## [89] tibble_3.1.4 KernSmooth_2.23-17
## [91] crayon_1.4.1 htmltools_0.5.2
## [93] mgcv_1.8-33 later_1.3.0
## [95] tidyr_1.1.3 DBI_1.1.1
## [97] ExperimentHub_1.16.1 dbplyr_2.1.1
## [99] MASS_7.3-53 rappdirs_0.3.3
## [101] Matrix_1.3-4 cli_3.0.1
## [103] igraph_1.2.6 pkgconfig_2.0.3
## [105] scuttle_1.0.4 plotly_4.9.4.1
## [107] spatstat.sparse_2.0-0 vipor_0.4.5
## [109] bslib_0.3.0 XVector_0.30.0
## [111] stringr_1.4.0 digest_0.6.27
## [113] sctransform_0.3.2 RcppAnnoy_0.0.19
## [115] spatstat.data_2.1-0 rmarkdown_2.10
## [117] leiden_0.3.9 uwot_0.1.10
## [119] DelayedMatrixStats_1.12.3 curl_4.3.2
## [121] shiny_1.6.0 lifecycle_1.0.0
## [123] nlme_3.1-149 jsonlite_1.7.2
## [125] BiocNeighbors_1.8.2 viridisLite_0.4.0
## [127] limma_3.46.0 fansi_0.5.0
## [129] pillar_1.6.2 lattice_0.20-41
## [131] fastmap_1.1.0 httr_1.4.2
## [133] survival_3.2-7 interactiveDisplayBase_1.28.0
## [135] glue_1.4.2 png_0.1-7
## [137] BiocVersion_3.12.0 bit_4.0.4
## [139] stringi_1.7.4 sass_0.4.0
## [141] blob_1.2.2 BiocSingular_1.6.0
## [143] AnnotationHub_2.22.1 memoise_2.0.0
## [145] irlba_2.3.3 future.apply_1.8.1
可以看出,各种注释方法都是比较科学的,但最大的挑战来源于参考数据集与待注释数据集间的适用度。
OK 吃席!
本系列其他课程
手把手教你做单细胞测序数据分析(一)——绪论
手把手教你做单细胞测序数据分析(二)——各类输入文件读取
手把手教你做单细胞测序数据分析(三)——单样本分析
手把手教你做单细胞测序数据分析(四)——多样本整合
手把手教你做单细胞测序数据分析(五)——细胞类型注释
手把手教你做单细胞测序数据分析(六)——组间差异分析及可视化
手把手教你做单细胞测序数据分析(七)——基因集富集分析
欢迎关注同名公众号~
