R语言 使用getGEO()直接进行差异表达分析并显示Entrez_id和Symbol_id
引入
平常我们利用GEODatasets的表达量数据进行基因表达分析的时候,常常是下载表达矩阵,利用limma包进行分析,如果能找到注释包,那就把探针注释成Entrez_id或者是Symbol_id,如果找不到,那就下载对应GPL提供的注释文件,但是很可能还是不好注释,比如今天拿来做例子的GSE15222对应的GPL2700: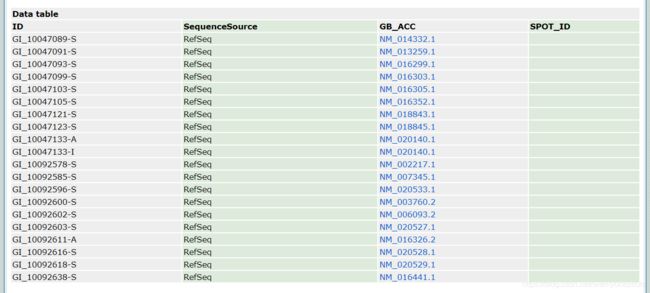
我们看到这个GPL只提供了GB_ACC也就是Genebank的一个号码,我们还要先将其转换成Entrez_id或者是Symbol_id(这个过程如果找不到好的工具很难完美地完成),然后利用Python或者Excel把这个GPL注释文件和下载的表达矩阵联系起来······如果你尝试着做一遍的话会发现非常消耗时间和精力。
但是,如果我们注意到GEO2R的话,会发现这个Series通过GEO Gatabase的在线分析工具GEO2R的分析之后,提供的结果是这个样子:
结果中竟然直接给了symbol_id!那么我们怎么才能得到这个呢?
GEO2R的运行脚本
GEO2R提供了分析的R脚本。
# Version info: R 3.2.3, Biobase 2.30.0, GEOquery 2.40.0, limma 3.26.8
# R scripts generated Tue Jan 15 02:03:41 EST 2019
################################################################
# Differential expression analysis with limma
library(Biobase)
library(GEOquery)
library(limma)
# load series and platform data from GEO
gset <- getGEO("GSE15222", GSEMatrix =TRUE, AnnotGPL=TRUE)
if (length(gset) > 1) idx <- grep("GPL2700", attr(gset, "names")) else idx <- 1
gset <- gset[[idx]]
# make proper column names to match toptable
fvarLabels(gset) <- make.names(fvarLabels(gset))
# group names for all samples
gsms <- paste0("XX000XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX",
"XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX",
"XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX",
"XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX111",
"1XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX",
"XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX",
"XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX",
"XXXXXXXXXXXXX")
sml <- c()
for (i in 1:nchar(gsms)) { sml[i] <- substr(gsms,i,i) }
# eliminate samples marked as "X"
sel <- which(sml != "X")
sml <- sml[sel]
gset <- gset[ ,sel]
# log2 transform
ex <- exprs(gset)
qx <- as.numeric(quantile(ex, c(0., 0.25, 0.5, 0.75, 0.99, 1.0), na.rm=T))
LogC <- (qx[5] > 100) ||
(qx[6]-qx[1] > 50 && qx[2] > 0) ||
(qx[2] > 0 && qx[2] < 1 && qx[4] > 1 && qx[4] < 2)
if (LogC) { ex[which(ex <= 0)] <- NaN
exprs(gset) <- log2(ex) }
# set up the data and proceed with analysis
sml <- paste("G", sml, sep="") # set group names
fl <- as.factor(sml)
gset$description <- fl
design <- model.matrix(~ description + 0, gset)
colnames(design) <- levels(fl)
fit <- lmFit(gset, design)
cont.matrix <- makeContrasts(G1-G0, levels=design)
fit2 <- contrasts.fit(fit, cont.matrix)
fit2 <- eBayes(fit2, 0.01)
tT <- topTable(fit2, adjust="fdr", sort.by="B", number=250)
tT <- subset(tT, select=c("ID","adj.P.Val","P.Value","t","B","logFC","Gene.symbol","Gene.title"))
write.table(tT, file=stdout(), row.names=F, sep="\t")
################################################################
# Boxplot for selected GEO samples
library(Biobase)
library(GEOquery)
# load series and platform data from GEO
gset <- getGEO("GSE15222", GSEMatrix =TRUE, getGPL=FALSE)
if (length(gset) > 1) idx <- grep("GPL2700", attr(gset, "names")) else idx <- 1
gset <- gset[[idx]]
# group names for all samples in a series
gsms <- paste0("XX000XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX",
"XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX",
"XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX",
"XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX111",
"1XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX",
"XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX",
"XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX",
"XXXXXXXXXXXXX")
sml <- c()
for (i in 1:nchar(gsms)) { sml[i] <- substr(gsms,i,i) }
sml <- paste("G", sml, sep="") set group names
# eliminate samples marked as "X"
sel <- which(sml != "X")
sml <- sml[sel]
gset <- gset[ ,sel]
# order samples by group
ex <- exprs(gset)[ , order(sml)]
sml <- sml[order(sml)]
fl <- as.factor(sml)
labels <- c("1","2")
# set parameters and draw the plot
palette(c("#dfeaf4","#f4dfdf", "#AABBCC"))
dev.new(width=4+dim(gset)[[2]]/5, height=6)
par(mar=c(2+round(max(nchar(sampleNames(gset)))/2),4,2,1))
title <- paste ("GSE15222", '/', annotation(gset), " selected samples", sep ='')
boxplot(ex, boxwex=0.6, notch=T, main=title, outline=FALSE, las=2, col=fl)
legend("topleft", labels, fill=palette(), bty="n")
分析这个R脚本我们能学到很多东西。首先,就是它的数据从哪里来?
在线获取表达矩阵
很明显,gset <- getGEO("GSE15222", GSEMatrix =TRUE, AnnotGPL=TRUE),这一句就直接获得了表达矩阵。我们按照bioconductor的方法安装好Biobase、GEOquery、limma三个包,运行这一句,得到gset是一个很复杂的列表: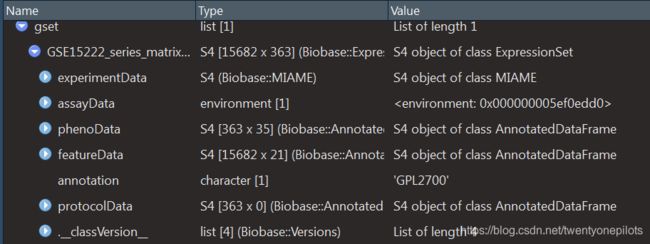
这个列表就是一个非常关键的数据。它不仅包括了我们可以下载的探针及其对应的表达量,还包括了Entrez_id和Symbol_id!
我们可以使用gset$GSE15222_series_matrix.txt.gz@featureData[[1]]显示探针数据;
用gset$GSE15222_series_matrix.txt.gz@featureData[[2]]显示基因的功能;
用gset$GSE15222_series_matrix.txt.gz@featureData[[3]]显示symbol_id;
用gset$GSE15222_series_matrix.txt.gz@featureData[[4]]显示entrez_id。
使用expr<-exprs(gset[[1]])获得我们需要的表达矩阵,它现在是这个样子: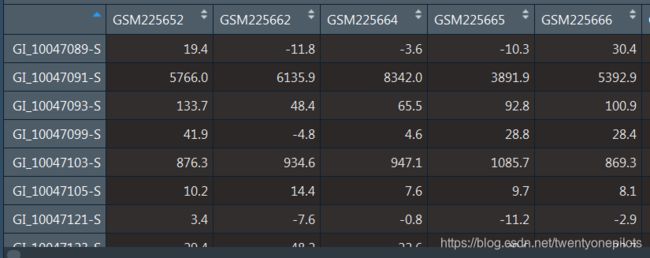
探针名现在是列名。
合并对应基因相同的探针
我们知道有一些探针并不对应基因,还有很多探针可能对应同一个基因。这要求我们合并对应基因相同的探针,删除不对应基因的探针。
在合并之前,先说一句Entrez_id和Symbol_id的关系。根据我的查询,Entrez_id对应Symbol_id是一个单射,一个Symbol_id可能对应多个Entrez_id。因此Entrez_id更为具体,我们合并探针直接将其转化为Entrez_id进行合并。那么这个时候为什么不直接在表达矩阵里注释上Symbol_id呢?这还要从合并方式谈起。
合并探针,我们使用的是函数aggregate(),有关于该函数的更多内容可以从R Studio内查询。
首先我们将entrez id直接作为一列插入到第一列前面(这也体现这种方法的方便性,对应关系已经给你弄好了):
entrez<-gset$GSE15222_series_matrix.txt.gz@featureData[[4]]
expr<-cbind(entrez,expr)
entrez=expr[,1]
现在expr是这个样子:

然后运行我们的合并并删除赘余的函数(调整参数可以选择合并方式,我这里选择的是根据entrez这个list进行合并,表达量合并时取平均值):
mydf<-aggregate(expr, by=list(entrez), FUN=mean)
合并之后我们获得的mydf是这样的(由于这个系列样本较多,因此运行时间较长,需要等几分钟):
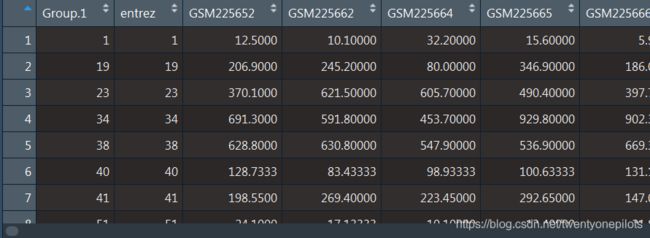
现在,这个数据框是按照entrez这列排的序,前面呢,多了一个Group.1这一列。我们接下来的任务是把entrez id放到列名,并且删去多余的这两列:
mydf <- subset(mydf, select = -entrez)
row.names(mydf)<-mydf$Group.1
mydf <- subset(mydf, select = -Group.1)
表达矩阵变成了这个样子:
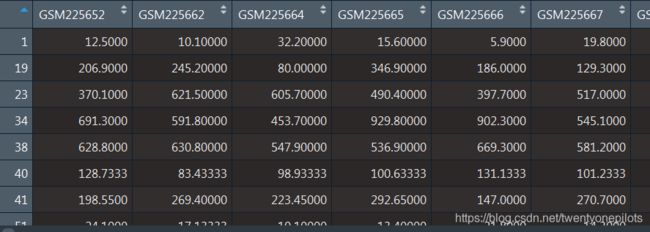
这时反过来想想为什么不能在之前加入Symbol_id。之前加入Symbol_id是很简单,直接插入这一列就好了。但是aggregate()函数并不能处理字符串,会把他们当成NA,所以加入Symbol_id相当于白加了。那么现在加Symbol_id可以吗?还是不行。因为接下来我们进行差异表达分析的时候仍然不能出现非数字类型的数据。
进行差异表达分析
这一步应该不用讲太多,算是很基础的一步了。
首先生成设计矩阵:
type <-c('Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD')
design <- model.matrix(~ -1+factor(type,levels=c('Control','AD'),ordered=TRUE))
colnames(design) <- c('Control','AD')
rownames(design)=colnames(mydf)
然后借助limma包进行差异表达分析就好了:
fit=lmFit(mydf, design)
contrast.matrix=makeContrasts(ControlVSAD=Control-AD,levels=design)
fit2 = contrasts.fit(fit, contrast.matrix)
fit2 = eBayes(fit2)
results <- decideTests(fit2)
vennDiagram(results)
#vennDiagram(object, include="both", names, mar=rep(1,4), cex=1.5, lwd=1, circle.col, counts.col, show.include, ...)
diff1 = topTreat(fit2, coef=1,p.value=0.05, n=Inf, adjust.method='BH')
此时的diff1是这个样子,按照调整后的P值排序的表,行名是Entrez_id:

添加Symbol_id
终于可以添加Symbol_id了。但是我们知道这个数据已经被打乱了,不仅探针合并、删除,最后的结果还是按照调整后的P值排序的。怎么让Symbol_id在diff1中对应上呢?那就要动用apply()函数了。
刚开始读入数据之后,我们首先创建一个id之间的对应字典:
symbol_id<-gset$GSE15222_series_matrix.txt.gz@featureData[[3]]
entrez<-gset$GSE15222_series_matrix.txt.gz@featureData[[4]]
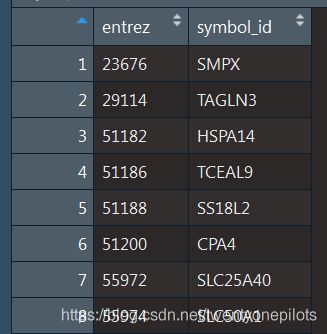
id<-cbind(entrez,symbol_id)
id是这样子的一个表:
获得diff1后,我们将diff1的行名,也就是Entrez_id提取出来作为一个数据框(列表),这样我们就获得了Entrez_id的顺序:
rn<-vector()
for(i in row.names(diff1)) rn<-c(rn,i)
rn<-as.data.frame(rn)
rn就是列名,接下来使用apply()函数,将id中的symbol_id以entrez作为字典中的"key"对应rn:
SYMBOL <-apply(
rn,
1, # 1 by row, 2 by column
function(x) id[which(id[,1]==x[1]),2][1] #如果最后不取[1](第一个元素),能合并的不同探针对应多个相同的entrez会重复出现在SYMBOL的一个位置
)
获得SYMBOL:

然后把它加到diff1的第一列即可:
diff1<-cbind(SYMBOL,diff1)
diff1就是最终我们需要的矩阵(如果需要基因信息也可以将基因信息加入),也可以将其导出为TXT:

源代码
最后贴上分析GSE15222的所有代码:
# Differential expression analysis with limma
library(Biobase)
library(GEOquery)
library(limma)
# load series and platform data from GEO
gset <- getGEO("GSE15222", GSEMatrix =TRUE, AnnotGPL=TRUE)
expr<-exprs(gset[[1]])
probe<-gset$GSE15222_series_matrix.txt.gz@featureData[[1]]
symbol_id<-gset$GSE15222_series_matrix.txt.gz@featureData[[3]]
entrez<-gset$GSE15222_series_matrix.txt.gz@featureData[[4]]
expr<-cbind(entrez,expr)
id<-cbind(entrez,symbol_id)
entrez=expr[,1]
mydf<-aggregate(expr, by=list(entrez), FUN=mean)
mydf <- subset(mydf, select = -entrez)
row.names(mydf)<-mydf$Group.1
mydf <- subset(mydf, select = -Group.1)
type <-c('Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','Control','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD','AD')
design <- model.matrix(~ -1+factor(type,levels=c('Control','AD'),ordered=TRUE))
colnames(design) <- c('Control','AD')
rownames(design)=colnames(mydf)
fit=lmFit(mydf, design)
contrast.matrix=makeContrasts(ControlVSAD=Control-AD,levels=design)
fit2 = contrasts.fit(fit, contrast.matrix)
fit2 = eBayes(fit2)
results <- decideTests(fit2)
vennDiagram(results)
#vennDiagram(object, include="both", names, mar=rep(1,4), cex=1.5, lwd=1, circle.col, counts.col, show.include, ...)
diff1 = topTreat(fit2, coef=1,p.value=0.05, n=Inf, adjust.method='BH')
rn<-vector()
for(i in row.names(diff1)) rn<-c(rn,i)
rn<-as.data.frame(rn)
SYMBOL <-apply(
rn,
1, # 1 by row, 2 by column
function(x) id[which(id[,1]==x[1]),2][1]
)
diff1<-cbind(SYMBOL,diff1)
write.table(diff1, "diff.ControlVSAD.GSE15222.txt",sep = '\t',quote = F)