生信技能-高通量测序工具bam、samtools、bedtools及conda的下载和安装
一、BWA
1、介绍
简介:用于建立 index;基于 BWT 算法,将 reads 比对到参考基因组;最新版本 bwa-mem2,Intel实验室对计算效率进行了优化。
详情:baw是一款将序列比对到参考基因组上的软件,用于高通量测序数据处理,包含了BWA-backtrack、BWA-SW、BWA-MEM三种算法:
1、BWA-backtrack:适合比对长度不超过100bp的序列;
2、BWA-SW和BWA-MEM适合于长度为70-1M bp的序列;其中BWA-MEM是最新开发的算法,对于高质量的测序数据,其比对的速度更快,精确度更高,对于70-100bp的reads, BWA-MEM算法在比对长度为70-100bp的序列时,效果比BWA-backtrack 算法的效果更好;
总而言之,通常情况下,选择BWA-MEM算法就好。
更多介绍请参考:https://bio-bwa.sourceforge.net/bwa.shtml
2、下载
bam下载地址
3、安装
打开终端,找到压缩文件所在位置;
# 安装
tar -jxvf bwa-*.tar.bz2
# 编译
cd bwa-0.7.17 # 要先进入对应的目录中
make bwa
# 添加环境变量
vim ~/.bash_profile # 编辑环境变量文件
export PATH=/Users/jolie/Desktop/工作/99-安装包/生信/bwa-0.7.17:$PATH # 编辑环境变量文件内容,文件所在路径要更新为你自己的地址哦!
# 使环境变量生效
source ~/.bashrc
# 验证是否安装成功
cd bwa-0.7.17 #如果添加了环境变量,在任意位置都可以执行,如果没有添加环境变量,则只能在对应目录下执行
bwa
二、Samtools
1、介绍
samtools是一个用于操作sam和bam文件的工具合集,包含有许多命令,同样用于用于高通量测序数据处理。
更多介绍请参考 http://samtools.sourceforge.net/samtools.shtml
2、下载
Samtools下载地址
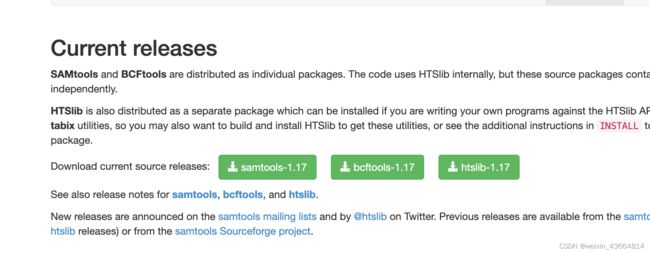
3、安装
打开终端,找到压缩文件所在位置;
# 安装
$ tar -jxvf samtools-*.tar.bz2
# 编译
cd samtools-1.17 # 要先进入对应的目录中
make
# 添加环境变量
vim ~/.bash_profile # 编辑环境变量文件
export PATH=/Users/jolie/Desktop/工作/99-安装包/生信/samtools-1.17:$PATH # 文件所在路径要更新为你自己的地址哦
# 使环境变量生效
source ~/.bashrc
# 验证是否安装成功
cd samtools-1.17 #如果添加了环境变量,在任意位置都可以执行,如果没有添加环境变量,则只能在对应目录下执行
samtools
4、使用
4.1、 view
1)、主要功能
将sam文件转换成bam文件;然后对bam文件进行各种操作。比如数据的排序(不属于本命令的功能)和提取(这些操作 是对bam文件进行的,因而当输入为sam文件的时候,不能进行该操作);最后将排序或提取得到的数据输出为bam或sam(默认的)格式。
2)、bam文件优点
bam文件为二进制文件,占用的磁盘空间比sam文本文件小;利用bam二进制文件的运算速度快。
3)、相关参数
- VIEW
view命令中,对sam文件头部的输入(-t或-T)和输出(-h)是单独的一些参数来控制的。
Usage: samtools view [options] <in.bam>|<in.sam> [region1 [...]]
# 默认情况下不加 region,则是输出所有的 region.
Options: -b output BAM
默认下输出是 SAM 格式文件,该参数设置输出 BAM 格式
-h print header for the SAM output
默认下输出的 sam 格式文件不带 header,该参数设定输出sam文件时带 header 信息
-H print header only (no alignments)
-S input is SAM
默认下输入是 BAM 文件,若是输入是 SAM 文件,则最好加该参数,否则有时候会报错。
-u uncompressed BAM output (force -b)
该参数的使用需要有-b参数,能节约时间,但是需要更多磁盘空间。
-c Instead of printing the alignments, only count them and print the
total number. All filter options, such as ‘-f’, ‘-F’ and ‘-q’ ,
are taken into account.
-1 fast compression (force -b)
-x output FLAG in HEX (samtools-C specific)
-X output FLAG in string (samtools-C specific)
-c print only the count of matching records
-L FILE output alignments overlapping the input BED FILE [null]
-t FILE list of reference names and lengths (force -S) [null]
使用一个list文件来作为header的输入
-T FILE reference sequence file (force -S) [null]
使用序列fasta文件作为header的输入
-o FILE output file name [stdout]
-R FILE list of read groups to be outputted [null]
-f INT required flag, 0 for unset [0]
-F INT filtering flag, 0 for unset [0]
Skip alignments with bits present in INT [0]
数字4代表该序列没有比对到参考序列上
数字8代表该序列的mate序列没有比对到参考序列上
-q INT minimum mapping quality [0]
-l STR only output reads in library STR [null]
-r STR only output reads in read group STR [null]
-s FLOAT fraction of templates to subsample; integer part as seed [-1]
-? longer help
4)、使用举例
# 将sam文件转换成bam文件
$ samtools view -bS abc.sam > abc.bam
$ samtools view -b -S abc.sam -o abc.bam
# 提取比对到参考序列上的比对结果
$ samtools view -bF 4 abc.bam > abc.F.bam
# 提取paired reads中两条reads都比对到参考序列上的比对结果,只需要把两个4+8的值12作为过滤参数即可
$ samtools view -bF 12 abc.bam > abc.F12.bam
# 提取没有比对到参考序列上的比对结果
$ samtools view -bf 4 abc.bam > abc.f.bam
# 提取bam文件中比对到caffold1上的比对结果,并保存到sam文件格式
$ samtools view abc.bam scaffold1 > scaffold1.sam
# 提取scaffold1上能比对到30k到100k区域的比对结果
$ samtools view abc.bam scaffold1:30000-100000 $gt; scaffold1_30k-100k.sam
# 根据fasta文件,将 header 加入到 sam 或 bam 文件中
$ samtools view -T genome.fasta -h scaffold1.sam > scaffold1.h.sam
4.2、 sort
1)、主要功能
sort对bam文件进行排序。
2)、相关参数
Usage: samtools sort [-n] [-m <maxMem>] <in.bam> <out.prefix>
-m 参数默认下是 500,000,000 即500M(不支持K,M,G等缩写)。对于处理大数据时,如果内存够用,则设置大点的值,以节约时间。
-n 设定排序方式按short reads的ID排序。默认下是按序列在fasta文件中的顺序(即header)和序列从左往右的位点排序。
3)、使用举例
$ samtools sort abc.bam abc.sort ###注意 abc.sort 是输出文件的前缀,实际输出是 abc.sort.bam
$ samtools view abc.sort.bam | less -S
4.3、 merge
1)、主要功能
将2个或2个以上的已经sort了的bam文件融合成一个bam文件。融合后的文件不需要则是已经sort过了的。
2)、相关参数
Usage: samtools merge [-nr] [-h inh.sam] <out.bam> <in1.bam> <in2.bam>[...]
# Samtools' merge does not reconstruct the @RG dictionary in the header. Users must provide the correct header with -h, or uses Picard which properly maintains the header dictionary in merging.
Options: -n sort by read names
-r attach RG tag (inferred from file names)
-u uncompressed BAM output
-f overwrite the output BAM if exist
-1 compress level 1
-R STR merge file in the specified region STR [all]
-h FILE copy the header in FILE to <out.bam> [in1.bam]
4.4、index
1)、主要功能
⚠️ 必须对bam文件进行默认情况下的排序后,才能进行index。否则会报错。
⚠️ 建立索引后将产生后缀为.bai的文件,用于快速的随机处理。很多情况下需要有bai文件的存在,特别是显示序列比对情况下。比如samtool的tview命令就需要;gbrowse2显示reads的比对图形的时候也需要。
2)、相关参数
Usage: samtools index <in.bam> [out.index]
3)、使用举例
# 以下两种命令结果一样
$ samtools index abc.sort.bam
$ samtools index abc.sort.bam abc.sort.bam.bai
4.5、
1)、主要功能
2)、相关参数
3)、使用举例
更多参数使用可参考samtools使用方法参数
三、bedtools
1、介绍
bedtools是处理基因组信息分析的强大工具集合,同样用于高通量测序数据处理。
更多介绍请参考 https://bedtools.readthedocs.io/en/latest/index.html
2、下载
curl -OL https://github.com/arq5x/bedtools2/releases/download/v2.22.0/bedtools-2.22.0.tar.gz
3、安装
# 安装
tar zxvf bedtools-2.22.0.tar.gz
# 编译
cd bedtools2 # 要先进入对应的目录中
make
# 添加环境变量
vim ~/.bash_profile # 编辑环境变量文件
export PATH=/Users/jolie/Desktop/工作/99-安装包/生信/bedtools2:$PATH
# 使环境变量生效
source ~/.bashrc
# 验证是否安装成功
cd bedtools #如果添加了环境变量,在任意位置都可以执行,如果没有添加环境变量,则只能在对应目录下执行
bedtools
4、相关参数
flexible tools for genome arithmetic and DNA sequence analysis.
usage: bedtools <subcommand> [options]
The bedtools sub-commands include:
[ Genome arithmetic ]
intersect Find overlapping intervals in various ways.
求区域之间的交集,可以用来注释peak,计算reads比对到的基因组区域
不同样品的peak之间的peak重叠情况。
window Find overlapping intervals within a window around an interval.
closest Find the closest, potentially non-overlapping interval.
寻找最近但可能不重叠的区域
coverage Compute the coverage over defined intervals.
计算区域覆盖度
map Apply a function to a column for each overlapping interval.
genomecov Compute the coverage over an entire genome.
merge Combine overlapping/nearby intervals into a single interval.
合并重叠或相接的区域
cluster Cluster (but don't merge) overlapping/nearby intervals.
complement Extract intervals _not_ represented by an interval file.
获得互补区域
subtract Remove intervals based on overlaps b/w two files.
计算区域差集
slop Adjust the size of intervals.
调整区域大小,如获得转录起始位点上下游3 K的区域
flank Create new intervals from the flanks of existing intervals.
sort Order the intervals in a file.
排序,部分命令需要排序过的bed文件
random Generate random intervals in a genome.
获得随机区域,作为背景集
shuffle Randomly redistrubute intervals in a genome.
根据给定的bed文件获得随机区域,作为背景集
sample Sample random records from file using reservoir sampling.
spacing Report the gap lengths between intervals in a file.
annotate Annotate coverage of features from multiple files.
[ Multi-way file comparisons ]
multiinter Identifies common intervals among multiple interval files.
unionbedg Combines coverage intervals from multiple BEDGRAPH files.
[ Paired-end manipulation ]
pairtobed Find pairs that overlap intervals in various ways.
pairtopair Find pairs that overlap other pairs in various ways.
[ Format conversion ]
bamtobed Convert BAM alignments to BED (& other) formats.
bedtobam Convert intervals to BAM records.
bamtofastq Convert BAM records to FASTQ records.
bedpetobam Convert BEDPE intervals to BAM records.
bed12tobed6 Breaks BED12 intervals into discrete BED6 intervals.
[ Fasta manipulation ]
getfasta Use intervals to extract sequences from a FASTA file.
提取给定位置的FASTA序列
maskfasta Use intervals to mask sequences from a FASTA file.
nuc Profile the nucleotide content of intervals in a FASTA file.
[ BAM focused tools ]
multicov Counts coverage from multiple BAMs at specific intervals.
tag Tag BAM alignments based on overlaps with interval files.
[ Statistical relationships ]
jaccard Calculate the Jaccard statistic b/w two sets of intervals.
计算数据集相似性
reldist Calculate the distribution of relative distances b/w two files.
fisher Calculate Fisher statistic b/w two feature files.
[ Miscellaneous tools ]
overlap Computes the amount of overlap from two intervals.
igv Create an IGV snapshot batch script.
用于生成一个脚本,批量捕获IGV截图
links Create a HTML page of links to UCSC locations.
makewindows Make interval "windows" across a genome.
把给定区域划分成指定大小和间隔的小区间 (bin)
groupby Group by common cols. & summarize oth. cols. (~ SQL "groupBy")
分组结算,不只可以用于bed文件。
expand Replicate lines based on lists of values in columns.
split Split a file into multiple files with equal records or base pairs.
—————————————
原文链接:https://blog.csdn.net/qazplm12_3/article/details/79797594
5、bedtools intersect的使用
Find overlapping intervals in various ways 求区域之间的交集,可以用来注释peak,计算reads比对到的基因组区域不同样品的peak之间的peak重叠情况
# 语法
bedtools intersect [OPTIONS] -a <bed/gff/vcf/bam> -b <bed/gff/vcf/bam>
#注:-b 可以接多个文件
相关参数可参考bedtools之intersect命令参数
# 使用举例
#找到A和B文件中重叠的部分前5行
bedtools intersect -a cpg.bed -b exons.bed | head -5
chr1 29320 29370 CpG:_116
chr1 135124 135563 CpG:_30
chr1 327790 328229 CpG:_29
chr1 327790 328229 CpG:_29
chr1 327790 328229 CpG:_29
#-wa:A和B重叠的区间再加上a的剩余部分
#-wb:A和B重叠的区间再加上b的剩余部分
bedtools intersect -a cpg.bed -b exons.bed -wa -wb | head -5
chr1 28735 29810 CpG:_116 chr1 29320 29370 NR_024540_exon_10_0_chr1_29321_r 0 -
chr1 135124 135563 CpG:_30 chr1 134772 139696 NR_039983_exon_0_0_chr1_134773_r 0 -
chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028322_exon_2_0_chr1_324439_f 0 +
chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028325_exon_2_0_chr1_324439_f 0 +
chr1 327790 328229 CpG:_29 chr1 327035 328581 NR_028327_exon_3_0_chr1_327036_f 0 +
#-wo Write the original A and B entries plus the number of base pairs of overlap between the two features. Only A features with overlap are reported
bedtools intersect -a cpg.bed -b exons.bed -wo | head -5
chr1 28735 29810 CpG:_116 chr1 29320 29370 NR_024540_exon_10_0_chr1_29321_r 0 - 50
chr1 135124 135563 CpG:_30 chr1 134772 139696 NR_039983_exon_0_0_chr1_134773_r 0 - 439
chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028322_exon_2_0_chr1_324439_f 0 + 439
chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028325_exon_2_0_chr1_324439_f 0 + 439
chr1 327790 328229 CpG:_29 chr1 327035 328581 NR_028327_exon_3_0_chr1_327036_f 0 + 439
#-c For each entry in A, report the number of hits in B while restricting to -f. Reports 0 for A entries that have no overlap with B
bedtools intersect -a cpg.bed -b exons.bed -c | head
chr1 28735 29810 CpG:_116 1
chr1 135124 135563 CpG:_30 1
chr1 327790 328229 CpG:_29 3
chr1 437151 438164 CpG:_84 0
chr1 449273 450544 CpG:_99 0
chr1 533219 534114 CpG:_94 0
chr1 544738 546649 CpG:_171 0
chr1 713984 714547 CpG:_60 1
chr1 762416 763445 CpG:_115 10
chr1 788863 789211 CpG:_28 9
# 找到覆盖了最多外显子的CPG岛
bedtools intersect -a cpg.bed -b exons.bed -c | sort -k5,5nr | head -2
chrY 15591259 15591720 CpG:_33 77
chrUn_gl000228 70214 114054 CpG:_3259 72
bedtools intersect -a cpg.bed -b exons.bed -c | sort -k1,1 -k2,2nr | head -2
chr1 249200252 249200721 CpG:_58 2
chr1 249167408 249168010 CpG:_48 0
#找到A文件中没有重叠B的部分
Only report those entries in A that have no overlap in B
bedtools intersect -a cpg.bed -b exons.bed -v | head
chr1 437151 438164 CpG:_84
chr1 449273 450544 CpG:_99
chr1 533219 534114 CpG:_94
chr1 544738 546649 CpG:_171
chr1 801975 802338 CpG:_24
chr1 805198 805628 CpG:_50
chr1 839694 840619 CpG:_83
chr1 844299 845883 CpG:_153
chr1 912869 913153 CpG:_28
chr1 919726 919927 CpG:_15
#从注释文件中,选取启动子
cat hesc.chromHmm.bed | grep Promoter > promoters.bed
cat promoters.bed |head -3
chr1 27737 28537 2_Weak_Promoter
chr1 28537 30137 1_Active_Promoter
chr1 30137 30337 2_Weak_Promoter
# 找到跟每个exon最近的启动子 多的一列数值是-a 和 -b 两者最近的距离
bedtools closest -a exons.bed -b promoters.bed -d | head -2
chr1 11873 12227 NR_046018_exon_0_0_chr1_11874_f 0 + chr1 27737 28537 2_Weak_Promoter 15511
chr1 12612 12721 NR_046018_exon_1_0_chr1_12613_f 0 + chr1 27737 28537 2_Weak_Promoter 15017
# 以5Kb一个窗口把人类基因组以覆盖
bedtools makewindows -g genome.txt -w 50000 > windows.bed
cat windows.bed |head -3
chr1 0 50000
chr1 50000 100000
chr1 100000 150000
bedtools makewindows -g genome.txt -w 100000 > windows0.bed
cat windows0.bed |head -3
chr1 0 100000
chr1 100000 200000
chr1 200000 300000
# 显示cpg.bed中和exons.bed有重叠的intervals
bedtools intersect -a cpg.bed -b exons.bed
# 显示exons.bed中和cpg.bed有重叠的intervals
bedtools intersect -a exons.bed -b cpg.bed
# 同时显示重叠区域的A、B文件中的原始记录
bedtools intersect -a exons.bed -b cpg.bed -wa -wb
# 显示重叠区域的碱基数
bedtools intersect -a cpg.bed -b exons.bed -wo
# 显示每一个cpg.bed文件中的记录在exons.bed文件中的重叠记录数
bedtools intersect -a cpg.bed -b exons.bed -c
# cpg.bed文件中不和exons.bed任何intervals重叠的记录
bedtools intersect -a cpg.bed -b exons.bed -v
bedtools intersect -a cpg.bed -b exons.bed -wo
# 设定阈值,显示cpg.bed中intervals至少有50%序列和exons.bed中的重叠
bedtools intersect -a cpg.bed -b exons.bed -wo -f 0.50
# 多个文件的重叠区域
bedtools intersect -a cpg.bed -b gwas.bed exons.bed
bedtools intersect -a cpg.bed -b gwas.bed exons.bed -wa -wb -names gwas exon # 加上文件label
# sorted数据通过加-sorted参数,运行速度更快
time bedtools intersect -a exons.bed -b cpg.bed gwas.bed -sorted >>/dev/null
—————————————
原文链接:https://blog.csdn.net/sunchengquan/article/details/85031173
原文链接:https://blog.csdn.net/qq_27390023/article/details/125433158
6、bedtools merge的使用
Combine overlapping/nearby intervals into a single interval 合并重叠或相接的区域
# 语法
bedtools merge [OPTIONS] -i <bed/gff/vcf>
#注意:bedtools merge要求输入文件先排序
# 使用举例
# 排序,输入文件先按染色体排序,然后按起始位置排序。
sort -k1,1 -k2,2n test.bed >test.sorted.bed
# 显示最终的"合并 "区间
bedtools merge -i exons.bed | head -n 20
# 在计算导致每个新的 "合并 "区间的重叠区间的数量时,我们将 "计算 "第一列。
bedtools merge -i exons.bed -c 1 -o count | head -n 20
# 显示所有合并成新的"合并 "区间的重叠区间的第二行
bedtools merge -i exons.bed -c 2 -o collapse | head -n 20
# 合并距离不超过1000的区间,
bedtools merge -i exons.bed -d 1000 -c 1 -o count | head -20
# 合并距离不超过90区域,分别对第一列和第四列做不同的操作
bedtools merge -i exons.bed -d 90 -c 1,4 -o count,collapse | head -20
————————————————
原文链接:https://blog.csdn.net/qq_27390023/article/details/125433158
7、bedtools complement的使用
Extract intervals not represented by an interval file 获得互补区域
# 语法
bedtools complement [OPTIONS] -i <bed/gff/vcf> -g <genome>
#注:The genome file should tab delimited and structured as follows:
<chromName><TAB><chromSize>
# 使用举例
# genome.txt中,exons.bed没有的区间
bedtools complement -i exons.bed -g genome.txt
8、bedtools genomecov的使用
Compute the coverage of a feature file among a genome 合并重叠或相接的区域
# 语法
bedtools genomecov [OPTIONS] -i <bed/gff/vcf> -g <genome>
#注:需要排序好的文件
bedtools genomecov -i exons.bed -g genome.txt
# 使用举例
# 输出BEDGRAPH,计算intervals的depth
bedtools genomecov -i exons.bed -g genome.txt -bg | head -20
9、bedtools jaccard的使用
calculate Jaccard statistic b/w two feature files 计算数据集相似性
# 语法
bedtools jaccard [OPTIONS] -a <bed/gff/vcf> -b <bed/gff/vcf>
# 使用举例
# 计算相似度
bedtools jaccard -a cpg.bed -b exons.bed
10、bedtools coverage的使用
Returns the depth and breadth of coverage of features from B 计算区域覆盖度
# 语法
# 使用举例
bedtools coverage [OPTIONS] -a <bed/gff/vcf> -b <bed/gff/vcf>
bedtools coverage -a cpg.bed -b exons.bed
四、conda
1、介绍
Conda是在Windows、macOS和Linux上运行的开源软件包管理系统和环境管理系统。可以快速安装、运行和更新软件包及其依赖项。可以轻松地在本地计算机上的环境中创建,保存,加载和切换。它是为Python程序创建的,但可以打包和分发适用于任何语言的软件。
目前conda的发行版本分为anaconda、miniconda两种,安装了ananconda或miniconda的完整版,就默认安装了conda。anaconda会包含一些常用包的版本,miniconda则是精简版,两者安装均可。
2、下载
miniconda下载地址
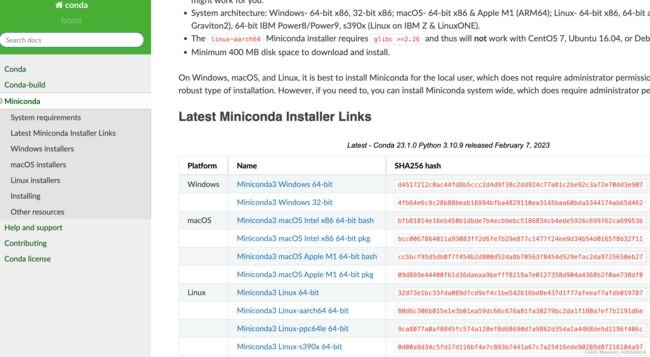
3、安装
# 进入miniconda所在目录
cd miniconda # 如果你的地址跟我不同,记得更新地址
# 执行安装命令
bash Miniconda3-py39_4.10.3-Linux-x86_64.sh # 如果你的版本跟我不同,记得更新名称

填写yes
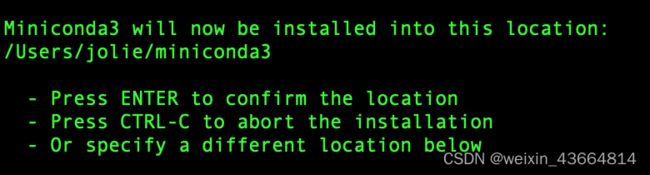
记得这里有选项是继续安装还是终止,继续安装即点击回车

填写yes

# 使环境变量生效
source ~/.bashrc
miniconda3安装成功~
⚠️ 安装成功后要关闭终端,再次进入才会生效哦~
# 添加环境变量
export PATH=/Users/jolie/miniconda3:$PATH # 文件所在路径要更新为你自己的地址哦
# 验证是否安装成功
cd miniconda3 #如果添加了环境变量,在任意位置都可以执行,如果没有添加环境变量,则只能在对应目录下执行
conda
# 查看相关帮助
conda -h # h参数为help的意思
4、conda管理packages
## 查看当前环境下的已经安装的包
conda list
## 查看频道下可用的某包
conda search openpyxl
## 安装包到环境work,不加--name时,默认安装到当前环境
conda install --name work openpyxl
## 安装包指定镜像源加速下载(**)
conda install -c https://mirrors.bfsu.edu.cn/anaconda/cloud/conda-forge micromamba
## 安装包requests , 默认路径下载很慢,此时可指定下载命令
conda install --name work -c conda-forge requests
## 检查安装结果
conda list
## 更新requests 包
conda update requests
## 卸载requests 包
conda remove requests
conda remove --name work requests # 删除work环境中的requests包
五、freebayes
1、介绍
简介:是一个常用于生物信息学数据分析中的免费开源软件,它用于从基因组或外显子组测序数据中检测SNP、indel和复合事件(例如插入或删除,或插入和SNP的组合)。该软件使用贝叶斯模型来计算每个位点的变异概率,并结合多个样本的测序数据来确定变异的共享情况。
优点:能够处理多个样本的测序数据,从而更好地确定遗传变异的共享情况。此外,它还可以处理多倍体或杂合基因组,并允许用户设置自定义的过滤标准来减少误报率。
应用:FreeBayes已被广泛用于各种生物信息学应用中,包括疾病基因组学研究、人类进化研究、农业遗传学研究和环境基因组学研究等领域。在分析大规模基因组测序数据时,FreeBayes是一种非常有用的工具,可以帮助研究人员鉴定遗传变异并进行精确的生物信息学分析。
2、下载
# 下载
git clone --recursive https://github.com/freebayes/freebayes.git
3、安装
# 依赖安装
# 在安装FreeBayes之前,需要先安装其所依赖的一些库和软件包。具体的依赖库可以在FreeBayes的GitHub仓库中找到。
# 在Ubuntu等Debian发行版中,可以使用以下命令安装依赖:
sudo apt-get install cmake git zlib1g-dev libbz2-dev liblzma-dev
# 在CentOS等Red Hat发行版中,可以使用以下命令安装依赖:
sudo yum install cmake git zlib-devel bzip2-devel xz-devel
# 如果在mac终端
/bin/bash -c "$(curl -fsSL https://raw.githubusercontent.com/Homebrew/install/HEAD/install.sh)" # 安装Homebrew,Homebrew为mac终端的软件包管理器
brew update #更新Homebrew
brew install freebayes #安装freebayes
# 编译
cd freebayes # 要先进入对应的目录中
make
# 添加环境变量
vim ~/.bash_profile # 编辑环境变量文件
export PATH=/Users/jolie/Desktop/工作/99-安装包/生信/freebayes:$PATH # 文件所在路径要更新为你自己的地址哦
# 使环境变量生效
source ~/.bashrc
# 验证是否安装成功
freebayes -h
4、相关参数
# 查看完整的freebayes参数列表和详细说明
freebayes --help