- Cell Insight | 单细胞测序技术又一新发现,可用于HIV-1和Mtb共感染个体诊断
尐尐呅
结核病是艾滋病合并其他疾病中导致患者死亡的主要原因。其中结核病由结核分枝杆菌(Mycobacteriumtuberculosis,Mtb)感染引起,获得性免疫缺陷综合症(艾滋病)由人免疫缺陷病毒(Humanimmunodeficiencyvirustype1,HIV-1)感染引起。国家感染性疾病临床医学研究中心/深圳市第三人民医院张国良团队携手深圳华大生命科学研究院吴靓团队,共同研究得出单细胞测序
- ArcGIS栅格计算器常见公式(赋值、0和空值的转换、补充栅格空值)
研学随笔
arcgis经验分享
我们在使用ArcGIS时通常经常用到栅格计算器,今天主要给大家介绍我日常中经常用到的几个公式,供大家参考学习。将特定值(-9999)赋值为0,例如-9999.Con("raster"==-9999,0,"raster")2.给空值赋予特定的值(如0)Con(IsNull("raster"),0,"raster")3.将特定的栅格值(如1)赋值为空值,其他保留原值SetNull("raster"==
- 网易严选官方旗舰店,优质商品,卓越服务
高省_飞智666600
网易严选官方旗舰店是网易旗下的一家电商平台,以提供优质商品和卓越服务而闻名。作为一名SEO优化师,我将为您详细介绍网易严选官方旗舰店,并重点强调其特点和优势。大家好!我是高省APP最大团队&联合创始人飞智导师。相较于其他返利app,高省APP的佣金更高,模式更好,最重要的是,终端用户不会流失!高省APP佣金更高,模式更好,终端用户不流失。【高省】是一个自用省钱佣金高,分享推广赚钱多的平台,百度有几
- 探索OpenAI和LangChain的适配器集成:轻松切换模型提供商
nseejrukjhad
langchaineasyui前端python
#探索OpenAI和LangChain的适配器集成:轻松切换模型提供商##引言在人工智能和自然语言处理的世界中,OpenAI的模型提供了强大的能力。然而,随着技术的发展,许多人开始探索其他模型以满足特定需求。LangChain作为一个强大的工具,集成了多种模型提供商,通过提供适配器,简化了不同模型之间的转换。本篇文章将介绍如何使用LangChain的适配器与OpenAI集成,以便轻松切换模型提供商
- 其二
十八尾喵
你知道吗?图片发自App我今天知道了你有喜欢的人,不是我。心空空的,整个人都不是我的了。可,怎么办?还是要好好的活着,毕竟你喜欢的人,我不能杀,可是我可以杀其他喜欢你的人呀!也罢,此生无缘,来世再见。鱼干
- 简介Shell、zsh、bash
zhaosuningsn
Shellzshbashshelllinuxbash
Shell是Linux和Unix的外壳,类似衣服,负责外界与Linux和Unix内核的交互联系。例如接收终端用户及各种应用程序的命令,把接收的命令翻译成内核能理解的语言,传递给内核,并把内核处理接收的命令的结果返回给外界,即Shell是外界和内核沟通的桥梁或大门。Linux和Unix提供了多种Shell,其中有种bash,当然还有其他好多种。Mac电脑中不但有bash,还有一个zsh,预装的,据说
- Python中深拷贝与浅拷贝的区别
yuxiaoyu.
转自:http://blog.csdn.net/u014745194/article/details/70271868定义:在Python中对象的赋值其实就是对象的引用。当创建一个对象,把它赋值给另一个变量的时候,python并没有拷贝这个对象,只是拷贝了这个对象的引用而已。浅拷贝:拷贝了最外围的对象本身,内部的元素都只是拷贝了一个引用而已。也就是,把对象复制一遍,但是该对象中引用的其他对象我不复
- 厉国刚:新闻学与传播学到底有何区别
微观大道
厉国刚:新闻学与传播学到底有何区别头几天,有人在知乎上问我:新闻学与传播学到底有何区别。他是一位想要跨专业考研的学生,对新闻传播学学科可谓了解甚少,甚至一头雾水,想要让我帮他解释解释。在研究生学硕层面,新闻传播学是一级学科,分成新闻学、传播学这两个二级学科。有些高校,还自设了广告学、出版发行学等其他二级学科,但从官方角度,新闻传播学一级学科下,正统的就是那两个二级学科。招生时,一般会按一级学科招,
- 简单了解 JVM
记得开心一点啊
jvm
目录♫什么是JVM♫JVM的运行流程♫JVM运行时数据区♪虚拟机栈♪本地方法栈♪堆♪程序计数器♪方法区/元数据区♫类加载的过程♫双亲委派模型♫垃圾回收机制♫什么是JVMJVM是JavaVirtualMachine的简称,意为Java虚拟机。虚拟机是指通过软件模拟的具有完整硬件功能的、运行在一个完全隔离的环境中的完整计算机系统(如:JVM、VMwave、VirtualBox)。JVM和其他两个虚拟机
- 关于Mysql 中 Row size too large (> 8126) 错误的解决和理解
秋刀prince
mysqlmysql数据库
提示:啰嗦一嘴,数据库的任何操作和验证前,一定要记得先备份!!!不会有错;文章目录问题发现一、问题导致的可能原因1、页大小2、行格式2.1compact格式2.2Redundant格式2.3Dynamic格式2.4Compressed格式3、BLOB和TEXT列二、解决办法1、修改页大小(不推荐)2、修改行格式3、修改数据类型为BLOB和TEXT列4、其他优化方式(可以参考使用)4.1合理设置数据
- 【从浅识到熟知Linux】Linux发展史
Jammingpro
从浅学到熟知Linuxlinux运维服务器
归属专栏:从浅学到熟知Linux个人主页:Jammingpro每日努力一点点,技术变化看得见文章前言:本篇文章记录Linux发展的历史,因在介绍Linux过程中涉及的其他操作系统及人物,本文对相关内容也有所介绍。文章目录Unix发展史Linux发展史开源Linux官网企业应用情况发行版本在学习Linux前,我们可能都会问Linux从哪里来?它是如何发展的。但在介绍Linux之前,需要先介绍一下Un
- 如何培养兴趣
绽蕊向阳
今天读李笑来的书《与时间做朋友》,读到有关兴趣部分,深有感触。书中提到,好多人说对某事没有兴趣,实际上是没有能力把这件事做好,做这件事时的感受很不好,有挫败感,每个人对自己不擅长做不好的事情,都本能的容易逃避,所以就以为自己对这件事不感兴趣,他们真正感兴趣的是其他事情。可事实上,出现这种感觉应该仅仅是因为还没有开始做那件事情,也还没有在那件事情上遭受挫折而已。其实,很多人真的放弃原来做的事情,转去
- 戴容容 中原焦点团队.网络初级第33期,坚持分享第19天 2022年3月9日
TessDai
《每个人眼中的世界都是不同的》“一千个人眼里有一千个哈姆雷特”世界是多元的,每个人都有自己的道理,人人按照自己的理解去看待这个世界的人和物.我们如此,其他人也是如此.因此,任何事情,我们要放下自己以为的真理,去理解他人认为的真理,只有同频方能共振.孩子在慢慢长大的过程中慢慢学会独立,甚至对抗.尤其当孩子处于青春期的时候,他们开始有很多自己独立的想法,和一些特立独行的做法,家长常常会觉得不可思议,觉
- 系统架构设计师 需求分析篇二
AmHardy
软件架构设计师系统架构需求分析面向对象分析分析模型UML和SysML
面向对象分析方法1.用例模型构建用例模型一般需要经历4个阶段:识别参与者:识别与系统交互的所有事物。合并需求获得用例:将需求分配给予其相关的参与者。细化用例描述:详细描述每个用例的功能。调整用例模型:优化用例之间的关系和结构,前三个阶段是必需的。2.用例图的三元素参与者:使用系统的用户或其他外部系统和设备。用例:系统所提供的服务。通信关联:参与者和用例之间的关系,或用例与用例之间的关系。3.识别参
- 每时每刻都是开始2019-03-09
Action熊猫
过去有多少想了无数遍要做的,但实际并没有做到的。以没时间,或其他种种自己认为可以接受的理由,看着一天走啦,一月去啦,又是一年。最后笑一笑,新年不是来了吗!重新开始...如果在过去的365天里,每次醒来,都没能开始,那新年来了,又如何呢?何不把人生的每时每刻都作为起点,不等待,不期盼,不自欺,让每时每刻都在开始中...。
- 2022-02-15
百味人生摆渡人
习惯她和他在谈恋时,就吵架,有时吵的还很厉害,不像其他恋人,恋爱期间有说不完的情话,享受不尽的温情,珍惜在一块的每一分每一秒。分手也分了无数次,不知怎的,就是没有分开。不见面时,特想见面,见了面时,说不了几句,又就开始吵,吵的次数多了,连他们自己也说不清值不值得吵,为什么吵。她妈妈说,这孩子从小就性格倔强。他爸爸说,这孩子从小就性格固执。想想其实也没什么大事。为吃什么吵为穿什么吵为说什么吵……日子
- android 更改窗口的层次,浮窗开发之窗口层级
Ms.Bu
android更改窗口的层次
最近在项目中遇到了这样的需求:需要在特定的其他应用之上悬浮自己的UI交互(拖动、输入等复杂的UI交互),和九游的浮窗类似,不过我们的比九游的体验更好,我们越过了很多授权的限制。浮窗效果很多人都知道如何去实现一个简单的浮窗,但是却很少有人去深入的研究背后的流程机制,由于项目中浮窗交互比较复杂,遇到了些坑查看了很多资料,故总结浮窗涉及到的知识点:窗口层级关系(浮窗是如何“浮”的)?浮窗有哪些限制,如何
- 06选课支付模块之基于消息队列发送支付通知消息
echo 云清
学成在线javarabbitmq消息队列支付通知学成在线
消息队列发送支付通知消息需求分析订单服务作为通用服务,在订单支付成功后需要将支付结果异步通知给其他对接的微服务,微服务收到支付结果根据订单的类型去更新自己的业务数据技术方案使用消息队列进行异步通知需要保证消息的可靠性即生产端将消息成功通知到服务端:消息发送到交换机-->由交换机发送到队列-->消费者监听队列,收到消息进行处理,参考文章02-使用Docker安装RabbitMQ-CSDN博客生产者确
- 基于STM32的汽车仪表显示系统:集成CAN、UART与I2C总线设计流程
极客小张
stm32汽车嵌入式硬件物联网单片机c语言
一、项目概述项目目标与用途本项目旨在设计和实现一个基于STM32微控制器的汽车仪表显示系统。该系统能够实时显示汽车的速度、转速、油量等关键信息,并通过CAN总线与其他汽车控制单元进行通信。这种仪表显示系统不仅提高了驾驶的安全性和便捷性,还能为汽车提供更智能的用户体验。技术栈关键词微控制器:STM32显示技术:TFTLCD/OLED传感器:速度传感器、温度传感器、油量传感器通信协议:CAN总线、UA
- 淘陶居老袁藏品
东海堂
【造像艺术】文化遗产•汉地木造像的区域特征、古代精品造像欣赏。。。。。。(来源:蠢牛/颜旭茂)原创2016-06-12作者:作者:蠢牛(颜旭茂)木造像的地位一直挺尴尬的。国外大型博物馆的木造像基本都是宋元以前的,明代只藏极品。国内也就故宫、国博和上博有能力弄几尊宋木,山西省博貌似只有一尊顶级的明代菩萨能拿得出手,其他木雕大省的博物馆再怎么也应当展示些明清木雕吧,总比同时代那什么坛坛罐罐更有艺术性。
- 【创客文案社】第三期写手招募
筱瑶123
创客文案社第三期写手招募开始了。要求:1:注册一个月以上2:本身热爱写作3:有时间参与接单投稿参与方式:可以关注公众号:写作灵感;也可以通过其他转发文章的文友帮忙拉入群;也可以简信我。参与之后的文友,会先进入新人班,进行基本的试稿与培训,先接一些比较简单的单子;在这里可以一边赚钱,一边学习。不知不觉,来三四个月了,也发现了很多很有意思的现象。1:在上写一篇文章,基本都是几毛钱,多的也不过几块钱的收
- 中国广电永久9元流量套餐!性价比最高流量卡套餐介绍!
优惠攻略官
中国广电是中国最大的传媒集团之一,其推出的流量套餐备受消费者青睐。中国广电最实惠的流量套餐不仅价格亲民,而且提供了优质的网络体验。首先,中国广电的流量套餐价格实惠,适合不同消费者的需求。无论是短期的日租卡还是长期有效的月租卡,用户都可以根据自己的实际情况选择适合自己的套餐。而且,流量的价格相对于其他运营商的套餐来说更加合理,给用户提供了更大的选择空间。☞大流量卡套餐「→点这免费申请办理」或者截图扫
- Python实现TIFF 文件转换为 PNG 和 JPG 格式
sand&wich
python开发语言
在日常的图像处理工作中,可能会遇到需要将TIFF格式的图像转换为其他格式的情况,例如PNG和JPG。下面,本文将介绍如何使用Python和GDAL库实现这一功能。准备工作在开始之前,请确保已经安装了必要的库:GDAL(GeospatialDataAbstractionLibrary)可以使用以下命令安装GDAL:pipinstallgdal代码实现以下是一个将TIFF文件转换为PNG文件的示例代码
- Python 课程10-单元测试
可愛小吉
Python教學python单元测试开发语言TDDunittest
前言在现代软件开发中,单元测试已成为一种必不可少的实践。通过测试,我们可以确保每个功能模块在开发和修改过程中按预期工作,从而减少软件缺陷,提高代码质量。而测试驱动开发(TDD)则进一步将测试作为开发的核心部分,先编写测试,再编写代码,以测试为指导开发出更稳定、更可靠的代码。Python提供了强大的unittest模块,它是Python标准库的一部分,专门用于编写和执行单元测试。与其他测试框架相比,
- 计算机毕业设计PHP仓储综合管理系统(源码+程序+VUE+lw+部署)
java毕设程序源码王哥
php课程设计vue.js
该项目含有源码、文档、程序、数据库、配套开发软件、软件安装教程。欢迎交流项目运行环境配置:phpStudy+Vscode+Mysql5.7+HBuilderX+Navicat11+Vue+Express。项目技术:原生PHP++Vue等等组成,B/S模式+Vscode管理+前后端分离等等。环境需要1.运行环境:最好是小皮phpstudy最新版,我们在这个版本上开发的。其他版本理论上也可以。2.开发
- Python编程 - 初识面向对象
易辰君
Python核心编程python开发语言
目录前言一、面向对象二、类和对象(一)类简介定义类(二)对象简介创建对象(三)总结三、实例属性和实例方法(一)实例属性创建的基本语法使用示例(二)实例方法定义实例方法的基本语法调用示例方法的示例(三)总结四、类中的self(一)基本概念(二)作用访问实例属性调用其他实例方法在构造函数中初始化对象(三)总结五、__init__方法(一)__init__方法的特点(二)基本语法(三)示例(四)总结前言
- [实践应用] 深度学习之模型性能评估指标
YuanDaima2048
深度学习工具使用深度学习人工智能损失函数性能评估pytorchpython机器学习
文章总览:YuanDaiMa2048博客文章总览深度学习之模型性能评估指标分类任务回归任务排序任务聚类任务生成任务其他介绍在机器学习和深度学习领域,评估模型性能是一项至关重要的任务。不同的学习任务需要不同的性能指标来衡量模型的有效性。以下是对一些常见任务及其相应的性能评估指标的详细解释和总结。分类任务分类任务是指模型需要将输入数据分配到预定义的类别或标签中。以下是分类任务中常用的性能指标:准确率(
- [实践应用] 深度学习之优化器
YuanDaima2048
深度学习工具使用pytorch深度学习人工智能机器学习python优化器
文章总览:YuanDaiMa2048博客文章总览深度学习之优化器1.随机梯度下降(SGD)2.动量优化(Momentum)3.自适应梯度(Adagrad)4.自适应矩估计(Adam)5.RMSprop总结其他介绍在深度学习中,优化器用于更新模型的参数,以最小化损失函数。常见的优化函数有很多种,下面是几种主流的优化器及其特点、原理和PyTorch实现:1.随机梯度下降(SGD)原理:随机梯度下降通过
- wandb一直上传 解决方案
行业边缘的摸鱼怪
bug解决方案服务器linux服务器
问题描述运行带有wandb的代码时,虽然可以实现及时同步非常方便,但当设置错参数或其他原因不得不使用ctrl+C停止运行时,总会出现wandb一直上传个不停的现象,给在同一终端重新运行新的代码造成困难。解决方案运行以下代码把wandb的进程直接杀死。psaux|grepwandb|grep-vgrep|awk'{print$2}'|xargskill-9参考链接[CLI]:Ctrl+Ctokill
- Python实现关联规则推荐
这孩子谁懂哈
PythonMachineLearningpython关联规则机器学习
1.什么关联规则关联规则(AssociationRules)是反映一个事物与其他事物之间的相互依存性和关联性,如果两个或多个事物之间存在一定的关联关系,那么,其中一个事物就能通过其他事物预测到。关联规则是数据挖掘的一个重要技术,用于从大量数据中挖掘出有价值的数据项之间的相关关系。关联规则挖掘的最经典的例子就是沃尔玛的啤酒与尿布的故事,通过对超市购物篮数据进行分析,即顾客放入购物篮中不同商品之间的关
- java封装继承多态等
麦田的设计者
javaeclipsejvmcencapsulatopn
最近一段时间看了很多的视频却忘记总结了,现在只能想到什么写什么了,希望能起到一个回忆巩固的作用。
1、final关键字
译为:最终的
&
- F5与集群的区别
bijian1013
weblogic集群F5
http请求配置不是通过集群,而是F5;集群是weblogic容器的,如果是ejb接口是通过集群。
F5同集群的差别,主要还是会话复制的问题,F5一把是分发http请求用的,因为http都是无状态的服务,无需关注会话问题,类似
- LeetCode[Math] - #7 Reverse Integer
Cwind
java题解MathLeetCodeAlgorithm
原题链接:#7 Reverse Integer
要求:
按位反转输入的数字
例1: 输入 x = 123, 返回 321
例2: 输入 x = -123, 返回 -321
难度:简单
分析:
对于一般情况,首先保存输入数字的符号,然后每次取输入的末位(x%10)作为输出的高位(result = result*10 + x%10)即可。但
- BufferedOutputStream
周凡杨
首先说一下这个大批量,是指有上千万的数据量。
例子:
有一张短信历史表,其数据有上千万条数据,要进行数据备份到文本文件,就是执行如下SQL然后将结果集写入到文件中!
select t.msisd
- linux下模拟按键输入和鼠标
被触发
linux
查看/dev/input/eventX是什么类型的事件, cat /proc/bus/input/devices
设备有着自己特殊的按键键码,我需要将一些标准的按键,比如0-9,X-Z等模拟成标准按键,比如KEY_0,KEY-Z等,所以需要用到按键 模拟,具体方法就是操作/dev/input/event1文件,向它写入个input_event结构体就可以模拟按键的输入了。
linux/in
- ContentProvider初体验
肆无忌惮_
ContentProvider
ContentProvider在安卓开发中非常重要。与Activity,Service,BroadcastReceiver并称安卓组件四大天王。
在android中的作用是用来对外共享数据。因为安卓程序的数据库文件存放在data/data/packagename里面,这里面的文件默认都是私有的,别的程序无法访问。
如果QQ游戏想访问手机QQ的帐号信息一键登录,那么就需要使用内容提供者COnte
- 关于Spring MVC项目(maven)中通过fileupload上传文件
843977358
mybatisspring mvc修改头像上传文件upload
Spring MVC 中通过fileupload上传文件,其中项目使用maven管理。
1.上传文件首先需要的是导入相关支持jar包:commons-fileupload.jar,commons-io.jar
因为我是用的maven管理项目,所以要在pom文件中配置(每个人的jar包位置根据实际情况定)
<!-- 文件上传 start by zhangyd-c --&g
- 使用svnkit api,纯java操作svn,实现svn提交,更新等操作
aigo
svnkit
原文:http://blog.csdn.net/hardwin/article/details/7963318
import java.io.File;
import org.apache.log4j.Logger;
import org.tmatesoft.svn.core.SVNCommitInfo;
import org.tmateso
- 对比浏览器,casperjs,httpclient的Header信息
alleni123
爬虫crawlerheader
@Override
protected void doGet(HttpServletRequest req, HttpServletResponse res) throws ServletException, IOException
{
String type=req.getParameter("type");
Enumeration es=re
- java.io操作 DataInputStream和DataOutputStream基本数据流
百合不是茶
java流
1,java中如果不保存整个对象,只保存类中的属性,那么我们可以使用本篇文章中的方法,如果要保存整个对象 先将类实例化 后面的文章将详细写到
2,DataInputStream 是java.io包中一个数据输入流允许应用程序以与机器无关方式从底层输入流中读取基本 Java 数据类型。应用程序可以使用数据输出流写入稍后由数据输入流读取的数据。
- 车辆保险理赔案例
bijian1013
车险
理赔案例:
一货运车,运输公司为车辆购买了机动车商业险和交强险,也买了安全生产责任险,运输一车烟花爆竹,在行驶途中发生爆炸,出现车毁、货损、司机亡、炸死一路人、炸毁一间民宅等惨剧,针对这几种情况,该如何赔付。
赔付建议和方案:
客户所买交强险在这里不起作用,因为交强险的赔付前提是:“机动车发生道路交通意外事故”;
如果是交通意外事故引发的爆炸,则优先适用交强险条款进行赔付,不足的部分由商业
- 学习Spring必学的Java基础知识(5)—注解
bijian1013
javaspring
文章来源:http://www.iteye.com/topic/1123823,整理在我的博客有两个目的:一个是原文确实很不错,通俗易懂,督促自已将博主的这一系列关于Spring文章都学完;另一个原因是为免原文被博主删除,在此记录,方便以后查找阅读。
有必要对
- 【Struts2一】Struts2 Hello World
bit1129
Hello world
Struts2 Hello World应用的基本步骤
创建Struts2的Hello World应用,包括如下几步:
1.配置web.xml
2.创建Action
3.创建struts.xml,配置Action
4.启动web server,通过浏览器访问
配置web.xml
<?xml version="1.0" encoding="
- 【Avro二】Avro RPC框架
bit1129
rpc
1. Avro RPC简介 1.1. RPC
RPC逻辑上分为二层,一是传输层,负责网络通信;二是协议层,将数据按照一定协议格式打包和解包
从序列化方式来看,Apache Thrift 和Google的Protocol Buffers和Avro应该是属于同一个级别的框架,都能跨语言,性能优秀,数据精简,但是Avro的动态模式(不用生成代码,而且性能很好)这个特点让人非常喜欢,比较适合R
- lua set get cookie
ronin47
lua cookie
lua:
local access_token = ngx.var.cookie_SGAccessToken
if access_token then
ngx.header["Set-Cookie"] = "SGAccessToken="..access_token.."; path=/;Max-Age=3000"
end
- java-打印不大于N的质数
bylijinnan
java
public class PrimeNumber {
/**
* 寻找不大于N的质数
*/
public static void main(String[] args) {
int n=100;
PrimeNumber pn=new PrimeNumber();
pn.printPrimeNumber(n);
System.out.print
- Spring源码学习-PropertyPlaceholderHelper
bylijinnan
javaspring
今天在看Spring 3.0.0.RELEASE的源码,发现PropertyPlaceholderHelper的一个bug
当时觉得奇怪,上网一搜,果然是个bug,不过早就有人发现了,且已经修复:
详见:
http://forum.spring.io/forum/spring-projects/container/88107-propertyplaceholderhelper-bug
- [逻辑与拓扑]布尔逻辑与拓扑结构的结合会产生什么?
comsci
拓扑
如果我们已经在一个工作流的节点中嵌入了可以进行逻辑推理的代码,那么成百上千个这样的节点如果组成一个拓扑网络,而这个网络是可以自动遍历的,非线性的拓扑计算模型和节点内部的布尔逻辑处理的结合,会产生什么样的结果呢?
是否可以形成一种新的模糊语言识别和处理模型呢? 大家有兴趣可以试试,用软件搞这些有个好处,就是花钱比较少,就算不成
- ITEYE 都换百度推广了
cuisuqiang
GoogleAdSense百度推广广告外快
以前ITEYE的广告都是谷歌的Google AdSense,现在都换成百度推广了。
为什么个人博客设置里面还是Google AdSense呢?
都知道Google AdSense不好申请,这在ITEYE上也不是讨论了一两天了,强烈建议ITEYE换掉Google AdSense。至少,用一个好申请的吧。
什么时候能从ITEYE上来点外快,哪怕少点
- 新浪微博技术架构分析
dalan_123
新浪微博架构
新浪微博在短短一年时间内从零发展到五千万用户,我们的基层架构也发展了几个版本。第一版就是是非常快的,我们可以非常快的实现我们的模块。我们看一下技术特点,微博这个产品从架构上来分析,它需要解决的是发表和订阅的问题。我们第一版采用的是推的消息模式,假如说我们一个明星用户他有10万个粉丝,那就是说用户发表一条微博的时候,我们把这个微博消息攒成10万份,这样就是很简单了,第一版的架构实际上就是这两行字。第
- 玩转ARP攻击
dcj3sjt126com
r
我写这片文章只是想让你明白深刻理解某一协议的好处。高手免看。如果有人利用这片文章所做的一切事情,盖不负责。 网上关于ARP的资料已经很多了,就不用我都说了。 用某一位高手的话来说,“我们能做的事情很多,唯一受限制的是我们的创造力和想象力”。 ARP也是如此。 以下讨论的机子有 一个要攻击的机子:10.5.4.178 硬件地址:52:54:4C:98
- PHP编码规范
dcj3sjt126com
编码规范
一、文件格式
1. 对于只含有 php 代码的文件,我们将在文件结尾处忽略掉 "?>" 。这是为了防止多余的空格或者其它字符影响到代码。例如:<?php$foo = 'foo';2. 缩进应该能够反映出代码的逻辑结果,尽量使用四个空格,禁止使用制表符TAB,因为这样能够保证有跨客户端编程器软件的灵活性。例
- linux 脱机管理(nohup)
eksliang
linux nohupnohup
脱机管理 nohup
转载请出自出处:http://eksliang.iteye.com/blog/2166699
nohup可以让你在脱机或者注销系统后,还能够让工作继续进行。他的语法如下
nohup [命令与参数] --在终端机前台工作
nohup [命令与参数] & --在终端机后台工作
但是这个命令需要注意的是,nohup并不支持bash的内置命令,所
- BusinessObjects Enterprise Java SDK
greemranqq
javaBOSAPCrystal Reports
最近项目用到oracle_ADF 从SAP/BO 上调用 水晶报表,资料比较少,我做一个简单的分享,给和我一样的新手 提供更多的便利。
首先,我是尝试用JAVA JSP 去访问的。
官方API:http://devlibrary.businessobjects.com/BusinessObjectsxi/en/en/BOE_SDK/boesdk_ja
- 系统负载剧变下的管控策略
iamzhongyong
高并发
假如目前的系统有100台机器,能够支撑每天1亿的点击量(这个就简单比喻一下),然后系统流量剧变了要,我如何应对,系统有那些策略可以处理,这里总结了一下之前的一些做法。
1、水平扩展
这个最容易理解,加机器,这样的话对于系统刚刚开始的伸缩性设计要求比较高,能够非常灵活的添加机器,来应对流量的变化。
2、系统分组
假如系统服务的业务不同,有优先级高的,有优先级低的,那就让不同的业务调用提前分组
- BitTorrent DHT 协议中文翻译
justjavac
bit
前言
做了一个磁力链接和BT种子的搜索引擎 {Magnet & Torrent},因此把 DHT 协议重新看了一遍。
BEP: 5Title: DHT ProtocolVersion: 3dec52cb3ae103ce22358e3894b31cad47a6f22bLast-Modified: Tue Apr 2 16:51:45 2013 -070
- Ubuntu下Java环境的搭建
macroli
java工作ubuntu
配置命令:
$sudo apt-get install ubuntu-restricted-extras
再运行如下命令:
$sudo apt-get install sun-java6-jdk
待安装完毕后选择默认Java.
$sudo update- alternatives --config java
安装过程提示选择,输入“2”即可,然后按回车键确定。
- js字符串转日期(兼容IE所有版本)
qiaolevip
TODateStringIE
/**
* 字符串转时间(yyyy-MM-dd HH:mm:ss)
* result (分钟)
*/
stringToDate : function(fDate){
var fullDate = fDate.split(" ")[0].split("-");
var fullTime = fDate.split("
- 【数据挖掘学习】关联规则算法Apriori的学习与SQL简单实现购物篮分析
superlxw1234
sql数据挖掘关联规则
关联规则挖掘用于寻找给定数据集中项之间的有趣的关联或相关关系。
关联规则揭示了数据项间的未知的依赖关系,根据所挖掘的关联关系,可以从一个数据对象的信息来推断另一个数据对象的信息。
例如购物篮分析。牛奶 ⇒ 面包 [支持度:3%,置信度:40%] 支持度3%:意味3%顾客同时购买牛奶和面包。 置信度40%:意味购买牛奶的顾客40%也购买面包。 规则的支持度和置信度是两个规则兴
- Spring 5.0 的系统需求,期待你的反馈
wiselyman
spring
Spring 5.0将在2016年发布。Spring5.0将支持JDK 9。
Spring 5.0的特性计划还在工作中,请保持关注,所以作者希望从使用者得到关于Spring 5.0系统需求方面的反馈。
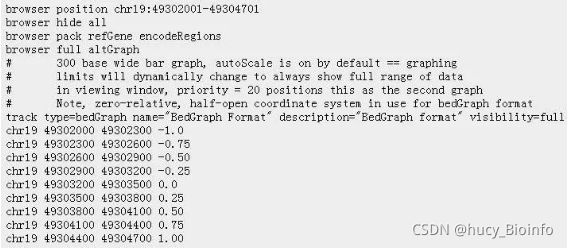
![NGS数据分析实践:03. 涉及的常用数据格式[4] - bed和Wiggle/Bigwig/bedgraph格式_第1张图片](http://img.e-com-net.com/image/info8/811c276e8e3b4f9fa44422c8071afedc.jpg)
![NGS数据分析实践:03. 涉及的常用数据格式[4] - bed和Wiggle/Bigwig/bedgraph格式_第2张图片](http://img.e-com-net.com/image/info8/5febb0e08de8493382d522445cd50271.jpg)
![NGS数据分析实践:03. 涉及的常用数据格式[4] - bed和Wiggle/Bigwig/bedgraph格式_第3张图片](http://img.e-com-net.com/image/info8/733ba1bffcf9437f8690d26845ff383f.jpg)
![NGS数据分析实践:03. 涉及的常用数据格式[4] - bed和Wiggle/Bigwig/bedgraph格式_第4张图片](http://img.e-com-net.com/image/info8/a0a82ee29e7341aca5bc0d56270c6d51.jpg)
![NGS数据分析实践:03. 涉及的常用数据格式[4] - bed和Wiggle/Bigwig/bedgraph格式_第5张图片](http://img.e-com-net.com/image/info8/6f7da5529ccb4f64a80f3557d1aeaf3a.jpg)