数据分析:转录组差异分析总结(DESeq2+limma+edgeR+t-test/wilcox-test)
前言
差异分析是转录组数据分析的必需技能之一,但众多的转录组分析R包如DESeq2,limma和edgeR等让分析人员不知如何选择,还有它们之间的优劣如何?我将在本文详细探讨常用差异分析R包以及结合t-test/wilcox-rank-sum test的差异分析结果的异同点。
大纲
本文要点由以下几点构成:
-
下载以及导入测试数据(批量安装R包);
-
基因表达count矩阵的标准化方法(F®PKM/TPM);
-
基因整体水平分布(PCA/tSNE/UMAP;heatmap);
-
DESeq2差异分析实现以及结果解析;
-
limma差异分析实现以及结果解析;
-
edgeR差异分析实现以及结果解析;
-
结合t-test或wilcox-rank-sum-test方法的差异分析实现以及结果解析(是否符合正态分布选择检验方法);
-
不同方法的结果比较(volcano plot+heatmap+venn);
-
总结。
导入R包
本次分析需要在R中批量安装包。先导入基础R包,在后面每个差异分析模块再导入所需要的差异分析R包。
library(dplyr)
library(tibble)
library(data.table)
library(ggplot2)
library(patchwork)
library(cowplot)
# rm(list = ls())
options(stringsAsFactors = F)
options(future.globals.maxSize = 1000 * 1024^2)
grp <- c("Normal", "Tumor")
grp.col <- c("#568875", "#73FAFC")
转录组数据
本文下载的TCGA-HNSC转录组数据是通过本人先前撰写的R脚本实现的,大家通过以下链接下载:
- 百度网盘链接:https://pan.baidu.com/s/178NlWqOa7mrl2QYq48N4WA
- 提取码: 4tth
phenotype <- fread("TCGA-HNSC-post_mRNA_clinical.csv")
count <- fread("TCGA-HNSC-post_mRNA_profile.tsv")
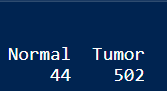
table(phenotype$Group)
标准化
标准化的目的是为了降低测序深度以及基因长度对基因表达谱下游分析的影响。测序深度越高则map到基因的reads也越多,同理基因长度越长则map到的reads也越多,最后对应的counts数目也越多。
- RPM/CPM: Reads/Counts of exon model per Million mapped reads (每百万映射读取的reads)
R P M = E x o n M a p p e d R e a d s ∗ 1 0 6 T o t a l M a p p e d R e a d s RPM = \frac{ExonMappedReads * 10^{6}}{TotalMappedReads} RPM=TotalMappedReadsExonMappedReads∗106
- RPKM: Reads Per Kilobase of exon model per Million mapped reads (每千个碱基的转录每百万映射读取的reads)
R P K M = E x o n M a p p e d R e a d s ∗ 1 0 9 T o t a l M a p p e d R e a d s ∗ E x o n L e n g t h RPKM = \frac{ExonMappedReads * 10^{9}}{TotalMappedReads * ExonLength} RPKM=TotalMappedReads∗ExonLengthExonMappedReads∗109
- FPKM: Fragments Per Kilobase of exon model per Million mapped fragments(每千个碱基的转录每百万映射读取的fragments), 适用于PE测序。
F P K M = E x o n M a p p e d F r a g m e n t s ∗ 1 0 9 T o t a l M a p p e d F r a g m e n t s ∗ E x o n L e n g t h FPKM = \frac{ExonMappedFragments * 10^{9}}{TotalMappedFragments * ExonLength} FPKM=TotalMappedFragments∗ExonLengthExonMappedFragments∗109
- TPM:Transcripts Per Kilobase of exon model per Million mapped reads (每千个碱基的转录每百万映射读取的Transcripts)
T P M = N i / L i ∗ 1 0 6 s u m ( N 1 / L 1 + N 2 / L 2 + . . . + N j / L j + . . . + N n / L n ) TPM= \frac{N_i/L_i * 10^{6}}{sum(N_1/L_1+N_2/L_2+...+N_j/L_j+...+N_n/L_n)} TPM=sum(N1/L1+N2/L2+...+Nj/Lj+...+Nn/Ln)Ni/Li∗106
N i N_i Ni为比对到第i个exon的reads数; L i L_i Li为第i个exon的长度;sum()为所有 (n个)exon按长度进行标准化之后数值的和
- 获取gene length表:对Homo_sapiens.GRCh38.101版本数据处理获取gene length数据;human_gene_all.tsv是使用biomart包获取gene symbol和ensembleID的对应关系表。
geneLength <- fread("Homo_sapiens.GRCh38.101.genelength.tsv")
geneIdAll <- fread("human_gene_all.tsv")
geneIdLength <- geneIdAll %>% filter(transcript_biotype == "protein_coding") %>%
dplyr::select(ensembl_gene_id, external_gene_name) %>%
inner_join(geneLength, by = c("ensembl_gene_id"="V1")) %>%
dplyr::select(-ensembl_gene_id) %>%
dplyr::distinct() %>%
dplyr::rename(Length=V2)
geneIdLengthUniq <- geneIdLength[pmatch(count$Feature, geneIdLength$external_gene_name), ] %>%
filter(!is.na(Length)) %>%
arrange(external_gene_name)
count_cln <- count %>% filter(Feature%in%geneIdLengthUniq$external_gene_name) %>%
arrange(Feature) %>% column_to_rownames("Feature")
if(!any(geneIdLengthUniq$external_gene_name == rownames(count_cln))){
message("Order of GeneName is wrong")
}
gene_lengths <-geneIdLengthUniq$Length
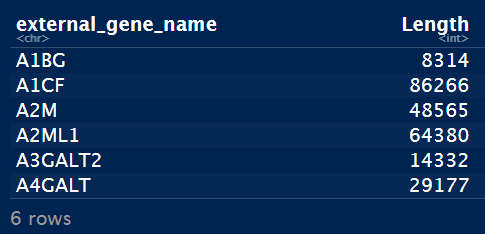
head(geneIdLengthUniq)
- RPKM/FPKM/TPM 转换
countToFpkm <- function(counts, lengths){
pm <- sum(counts) /1e6
rpm <- counts/pm
rpm/(lengths/1000)
}
countToTpm <- function(counts, lengths) {
rpk <- counts/(lengths/1000)
coef <- sum(rpk) / 1e6
rpk/coef
}
# FPKM
count_FPKM <- apply(count_cln, 2, function(x){countToFpkm(x, gene_lengths)}) %>%
data.frame()
# TPM
count_TPM <- apply(count_cln, 2, function(x){countToTpm(x, gene_lengths)}) %>%
data.frame()
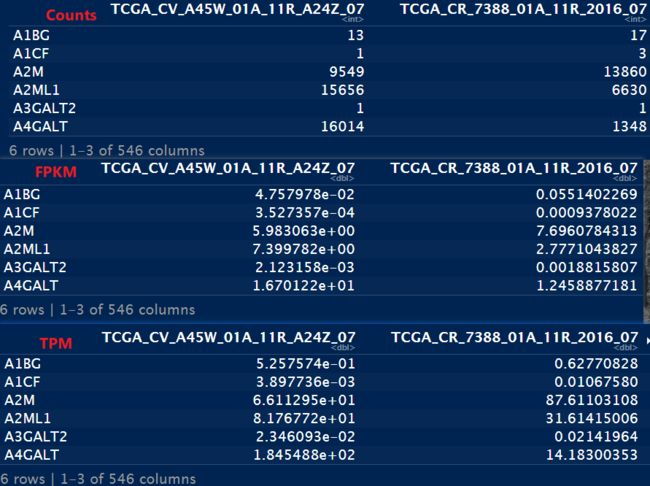
head(count_cln)
head(count_FPKM)
head(count_TPM)
整体水平比较
在做差异分析前,一般可以对数据做一个降维处理,然后看不同分组是否能在二维展开平面区分开。
ExpressionSet
先将数据存成ExpressionSet格式,可参考RNA-seq数据的批次校正方法文章。ExpressionSet对象数据包含表达谱和metadata等数据,这方便后期分析。
getExprSet <- function(metadata=phenotype,
profile=count_cln,
occurrence=0.2){
# metadata=phenotype
# profile=count_cln
# occurrence=0.2
sid <- intersect(metadata$SampleID, colnames(profile))
# phenotype
phe <- metadata %>% filter(SampleID%in%sid) %>%
column_to_rownames("SampleID")
# profile by occurrence
prf <- profile %>% rownames_to_column("tmp") %>%
filter(apply(dplyr::select(., -one_of("tmp")), 1, function(x) {
sum(x != 0)/length(x)}) > occurrence) %>%
dplyr::select(c(tmp, rownames(phe))) %>%
column_to_rownames("tmp")
# determine the right order between profile and phenotype
for(i in 1:ncol(prf)){
if (!(colnames(prf)[i] == rownames(phe)[i])) {
stop(paste0(i, " Wrong"))
}
}
require(convert)
exprs <- as.matrix(prf)
adf <- new("AnnotatedDataFrame", data=phe)
experimentData <- new("MIAME",
name="Hua Zou", lab="UCAS",
contact="[email protected]",
title="TCGA-HNSC",
abstract="The gene ExpressionSet",
url="www.zouhua.top",
other=list(notes="Created from text files"))
expressionSet <- new("ExpressionSet", exprs=exprs,
phenoData=adf,
experimentData=experimentData)
return(expressionSet)
}
ExprSet_count <- getExprSet(profile=count_cln)
ExprSet_FPKM <- getExprSet(profile=count_FPKM)
ExprSet_TPM <- getExprSet(profile=count_TPM)
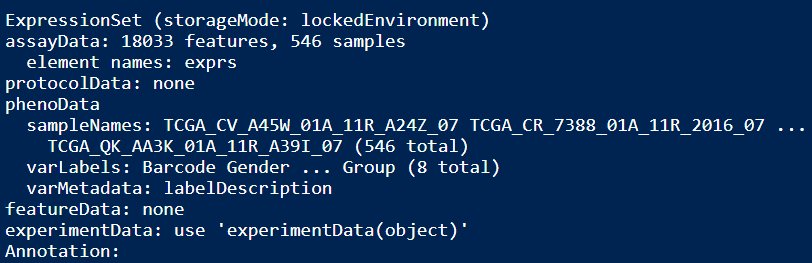
ExprSet_count
ExprSet_FPKM
ExprSet_TPM
降维分析
高纬度数据降维方法很多,我们这里选择了PCA+tSNE+UMAP分别展示降维后的结果,可参考文章高纬度数据降维方法的R实现。另外合并多个图的方法,请参考文章patchwork:合并多个R图的包。
- PCA
PCAFun <- function(dataset = ExprSet_count){
# dataset = ExprSet_count
require(convert)
metadata <- pData(dataset)
profile <- exprs(dataset)
# pca
pca <- prcomp(scale(t(profile), center = T, scale = T))
require(factoextra)
eig <- get_eig(pca)
# explains variable
explains <- paste0(paste0("PC", seq(2)), "(", paste0(round(eig[1:2, 2], 2), "%"), ")")
# principal component score of each sample
score <- inner_join(pca$x %>% data.frame() %>%
dplyr::select(c(1:2)) %>%
rownames_to_column("SampleID"),
metadata %>% rownames_to_column("SampleID"),
by = "SampleID") %>%
mutate(Group=factor(Group, levels = grp))
# PERMANOVA
require(vegan)
set.seed(123)
if(any(profile < 0)){
res_adonis <- adonis(vegdist(t(profile), method = "manhattan") ~ metadata$Group, permutations = 999)
}else{
res_adonis <- adonis(vegdist(t(profile), method = "bray") ~ metadata$Group, permutations = 999)
}
adn_pvalue <- res_adonis[[1]][["Pr(>F)"]][1]
adn_rsquared <- round(res_adonis[[1]][["R2"]][1],3)
#use the bquote function to format adonis results to be annotated on the ordination plot.
signi_label <- paste(cut(adn_pvalue,breaks=c(-Inf, 0.001, 0.01, 0.05, Inf), label=c("***", "**", "*", ".")))
adn_res_format <- bquote(atop(atop("PERMANOVA",R^2==~.(adn_rsquared)),
atop("p-value="~.(adn_pvalue)~.(signi_label), phantom())))
pl <- ggplot(score, aes(x=PC1, y=PC2))+
geom_point(aes(fill=Group), size=2, shape=21, stroke = .8, color = "black")+
stat_ellipse(aes(color=Group), level = 0.95, linetype = 1, size = 1.5)+
labs(x=explains[1], y=explains[2])+
scale_color_manual(values = grp.col)+
scale_fill_manual(name = "Condition",
values = grp.col)+
annotate("text", x = max(score$PC1) - 8,
y = min(score$PC1),
label = adn_res_format,
size = 6)+
guides(color="none")+
theme_classic()+
theme(axis.title = element_text(size = 10, color = "black", face = "bold"),
axis.text = element_text(size = 9, color = "black"),
text = element_text(size = 8, color = "black", family = "serif"),
strip.text = element_text(size = 9, color = "black", face = "bold"),
panel.grid = element_blank(),
legend.title = element_text(size = 11, color = "black", family = "serif"),
legend.text = element_text(size = 10, color = "black", family = "serif"),
legend.position = c(0, 0),
legend.justification = c(0, 0),
legend.background = element_rect(color = "black", fill = "white", linetype = 2, size = 0.5))
return(pl)
}
PCA_count <- PCAFun(dataset = ExprSet_count)
PCA_FPKM <- PCAFun(dataset = ExprSet_FPKM)
PCA_TPM <- PCAFun(dataset = ExprSet_TPM)
plot_grid(PCA_count, PCA_FPKM, PCA_TPM, nrow=1, align="hv", labels=c("Counts", "FPKM", "TPM"))
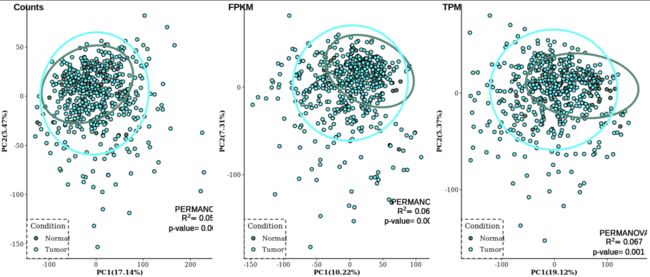
Notes: PCA的结果在三类数据中表现都不是很好,也就是组间区分不是明显,但是PERMANOVA的检验结果显示p值是显著差异的,可是 R 2 R^2 R2却偏低,也就是解释度很低。
- tSNE
RtsneFun <- function(dataset = ExprSet_count,
perpl = 30){
# dataset = ExprSet_count
# perpl = 10
require(convert)
metadata <- pData(dataset)
profile <- t(exprs(dataset))
# Rtsne
require(Rtsne)
#set.seed(123)
Rtsne <- Rtsne(profile,
dims=2,
perplexity=perpl,
verbose=TRUE,
max_iter=500,
eta=200)
point <- Rtsne$Y %>% data.frame() %>%
dplyr::select(c(1:2)) %>%
setNames(c("tSNE1", "tSNE2"))
rownames(point) <- rownames(profile)
score <- inner_join(point %>% rownames_to_column("SampleID"),
metadata %>% rownames_to_column("SampleID"),
by = "SampleID") %>%
mutate(Group=factor(Group, levels = grp))
# PERMANOVA
require(vegan)
set.seed(123)
if(any(profile < 0)){
res_adonis <- adonis(vegdist(profile, method = "manhattan") ~ metadata$Group, permutations = 999)
}else{
res_adonis <- adonis(vegdist(profile, method = "bray") ~ metadata$Group, permutations = 999)
}
adn_pvalue <- res_adonis[[1]][["Pr(>F)"]][1]
adn_rsquared <- round(res_adonis[[1]][["R2"]][1],3)
#use the bquote function to format adonis results to be annotated on the ordination plot.
signi_label <- paste(cut(adn_pvalue,breaks=c(-Inf, 0.001, 0.01, 0.05, Inf), label=c("***", "**", "*", ".")))
adn_res_format <- bquote(atop(atop("PERMANOVA",R^2==~.(adn_rsquared)),
atop("p-value="~.(adn_pvalue)~.(signi_label), phantom())))
pl <- ggplot(score, aes(x=tSNE1, y=tSNE2))+
geom_point(aes(fill=Group), size=3.5, shape=21, stroke = .8, color = "black")+
stat_ellipse(aes(color=Group), level = 0.95, linetype = 1, size = 1.5)+
scale_color_manual(values = grp.col)+
scale_fill_manual(name = "Condition",
values = grp.col)+
annotate("text", x = max(score$tSNE1) - 8,
y = max(score$tSNE2)-5,
label = adn_res_format,
size = 6)+
guides(color="none")+
theme_classic()+
theme(axis.title = element_text(size = 10, color = "black", face = "bold"),
axis.text = element_text(size = 9, color = "black"),
text = element_text(size = 8, color = "black", family = "serif"),
strip.text = element_text(size = 9, color = "black", face = "bold"),
panel.grid = element_blank(),
legend.title = element_text(size = 11, color = "black", family = "serif"),
legend.text = element_text(size = 10, color = "black", family = "serif"),
legend.position = c(0, 0),
legend.justification = c(0, 0),
legend.background = element_rect(color = "black", fill = "white", linetype = 2, size = 0.5))
return(pl)
}
Rtsne_count <- RtsneFun(dataset = ExprSet_count)
Rtsne_FPKM <- RtsneFun(dataset = ExprSet_FPKM)
Rtsne_TPM <- RtsneFun(dataset = ExprSet_TPM)
plot_grid(Rtsne_count, Rtsne_FPKM, Rtsne_TPM, nrow=1, align="hv", labels=c("Counts", "FPKM", "TPM"))
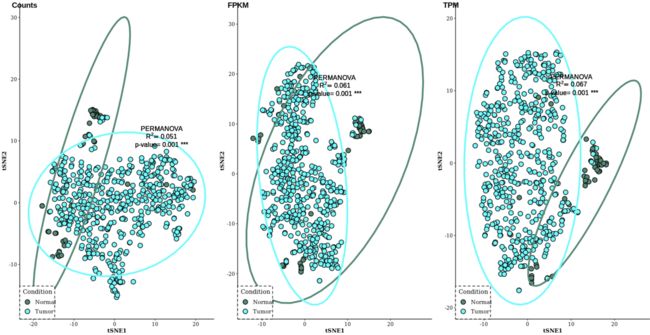
- UMAP
UMAPFun <- function(dataset = ExprSet_count){
# dataset = ExprSet_count
metadata <- pData(dataset)
profile <- t(exprs(dataset))
# umap
require(umap)
umap <- umap::umap(profile)
point <- umap$layout %>% data.frame() %>%
setNames(c("UMAP1", "UMAP2"))
rownames(point) <- rownames(profile)
score <- inner_join(point %>% rownames_to_column("SampleID"),
metadata %>% rownames_to_column("SampleID"),
by = "SampleID") %>%
mutate(Group=factor(Group, levels = grp))
# PERMANOVA
require(vegan)
set.seed(123)
if(any(profile < 0)){
res_adonis <- adonis(vegdist(profile, method = "manhattan") ~ metadata$Group, permutations = 999)
}else{
res_adonis <- adonis(vegdist(profile, method = "bray") ~ metadata$Group, permutations = 999)
}
adn_pvalue <- res_adonis[[1]][["Pr(>F)"]][1]
adn_rsquared <- round(res_adonis[[1]][["R2"]][1],3)
#use the bquote function to format adonis results to be annotated on the ordination plot.
signi_label <- paste(cut(adn_pvalue,breaks=c(-Inf, 0.001, 0.01, 0.05, Inf), label=c("***", "**", "*", ".")))
adn_res_format <- bquote(atop(atop("PERMANOVA",R^2==~.(adn_rsquared)),
atop("p-value="~.(adn_pvalue)~.(signi_label), phantom())))
pl <- ggplot(score, aes(x=UMAP1, y=UMAP2))+
geom_point(aes(fill=Group), size=2, shape=21, stroke = .8, color = "black")+
stat_ellipse(aes(color=Group), level = 0.95, linetype = 1, size = 1.5)+
scale_color_manual(values = grp.col)+
scale_fill_manual(name = "Condition",
values = grp.col)+
annotate("text", x = max(score$UMAP1),
y = min(score$UMAP2),
label = adn_res_format,
size = 6)+
guides(color="none")+
theme_classic()+
theme(axis.title = element_text(size = 10, color = "black", face = "bold"),
axis.text = element_text(size = 9, color = "black"),
text = element_text(size = 8, color = "black", family = "serif"),
strip.text = element_text(size = 9, color = "black", face = "bold"),
panel.grid = element_blank(),
legend.title = element_text(size = 11, color = "black", family = "serif"),
legend.text = element_text(size = 10, color = "black", family = "serif"),
legend.position = c(0, 0),
legend.justification = c(0, 0),
legend.background = element_rect(color = "black", fill = "white", linetype = 2, size = 0.5))
return(pl)
}
UMAP_count <- UMAPFun(dataset = ExprSet_count)
UMAP_FPKM <- UMAPFun(dataset = ExprSet_FPKM)
UMAP_TPM <- UMAPFun(dataset = ExprSet_TPM)
(UMAP_count + UMAP_FPKM + UMAP_TPM) + plot_layout(nrow = 1, guides = "collect")

总结:综合上述三种聚类分析方法,后两种基于非线性模型的方法具有较好的区分样本效果,而且TPM的区分效果貌似比Count和FPKM标准化方法更好。我后面应该度降维方法做一个原理上的总结。
热图
heatFun <- function(datset=ExprSet_count){
# datset=ExprSet_count
pheno <- pData(datset) %>% data.frame() %>%
rownames_to_column("SampleID") %>%
mutate(Group=factor(Group, levels = grp)) %>%
arrange(Group) %>%
column_to_rownames("SampleID")
edata <- exprs(datset) %>% data.frame()
# scale data: z-score
scale_rows <- function (x) {
m = apply(x, 1, mean, na.rm = T)
s = apply(x, 1, sd, na.rm = T)
return((x - m)/s)
}
edata_scaled <- t(scale_rows(edata))
require(circlize)
col_fun <- colorRamp2(c(round(range(edata_scaled)[1]), 0,
round(range(edata_scaled)[2])),
c("blue", "white", "red"))
# row split
dat_status <- table(pheno$Group)
dat_status_number <- as.numeric(dat_status)
dat_status_name <- names(dat_status)
row_split <- c()
for (i in 1:length(dat_status_number)) {
row_split <- c(row_split, rep(i, dat_status_number[i]))
}
require(ComplexHeatmap)
pl <- Heatmap(
edata_scaled,
#col = col_fun,
cluster_rows = FALSE,
row_order = rownames(pheno),
show_column_names = FALSE,
show_row_names = FALSE,
row_names_gp = gpar(fontsize = 12),
row_names_side = "right",
row_dend_side = "left",
column_title = NULL,
heatmap_legend_param = list(
title = "Abundance",
title_position = "topcenter",
border = "black",
legend_height = unit(10, "cm"),
direction = "horizontal"),
row_split = row_split,
left_annotation = rowAnnotation(foo = anno_block(gp = gpar(fill = 2:4),
labels = grp,
labels_gp = gpar(col = "black", fontsize = 12))),
column_km = 2
)
return(pl)
}
Heat_count <- heatFun(datset = ExprSet_count)
Heat_FPKM <- heatFun(datset = ExprSet_FPKM)
Heat_TPM <- heatFun(datset = ExprSet_TPM)
Heat_count
Note: 19000+基因呈现出来的heatmap太大了,暂时展示counts矩阵的结果。从这图能看出还是有部分区块基因是存在富聚集的。
DESeq2
DESeq2包输入的数据需要是counts矩阵,它使用负二项分布广义线性模型处理测序深度影响。该包的原理请参考文章差异表达分析之Deseq2。
计算差异结果
-
DESeqDataSetFromMatrix构建DESeq函数所需要的包含count矩阵的数据对象;
-
DESeq函数进行差异分析。
DESeq2Fun <- function(datset=ExprSet_count){
# datset=ExprSet_count
profile <- exprs(datset)
metadata <- pData(datset)
sid <- intersect(rownames(metadata), colnames(profile))
colData <- metadata %>% rownames_to_column("SampleID") %>%
dplyr::select(SampleID, Group) %>%
filter(SampleID%in%sid) %>%
mutate(Group=factor(Group, levels = grp)) %>%
column_to_rownames("SampleID")
countData <- profile %>% data.frame() %>%
dplyr::select(all_of(sid)) %>%
as.matrix()
if(!all(rownames(colData) == colnames(countData))){
stop("Order is wrong please check your data")
}
ddsm <- DESeqDataSetFromMatrix(countData = countData,
colData = colData,
design = ~ Group)
dds <- DESeq(ddsm)
return(dds)
}
DESeq_dds <- DESeq2Fun(datset=ExprSet_count)
DESeq_dds

Notes: DESeq2_1.28.1更新了构建DESeqDataSetFromMatrix函数,原来需要将feature单独成一列,现在则不需要了,但需要保障colData的行名和countData的列名保持一致并且都是样本名字。
提取结果
DESeq_result <- results(DESeq_dds, contrast = c("Group", rev(grp)))
![]()
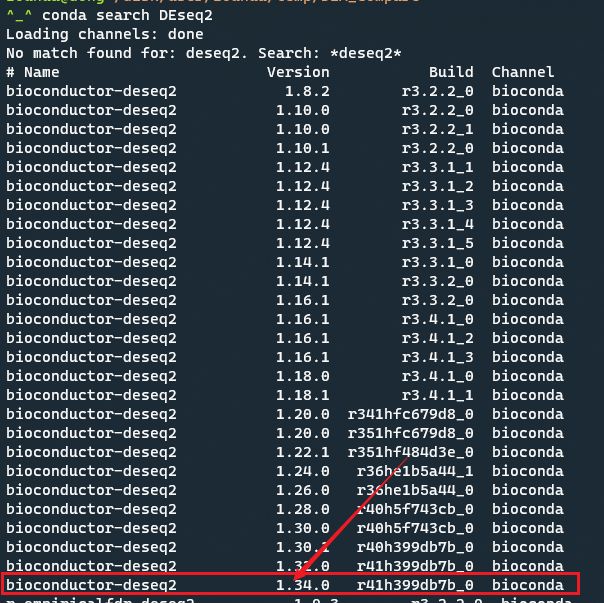
按照教程运行报错了,后来才发现是DESeq2_1.28.1版本的问题,更新到更高级的版本可解决该问题。这里因为是在Linux Rstudio server分析的,为了快捷安装我采用了conda模式。
conda search DEseq2
conda install -c bioconda bioconductor-deseq2=1.30.0 -y
也可以通过BiocManager::install安装
remove.packages("DESeq2")
BiocManager::install("DESeq2")
library(DESeq2)
在安装完成后,我们重新运行上述R代码,提取出DESeq2的结果文件。
DESeq_dds <- DESeq2Fun(datset=ExprSet_count)
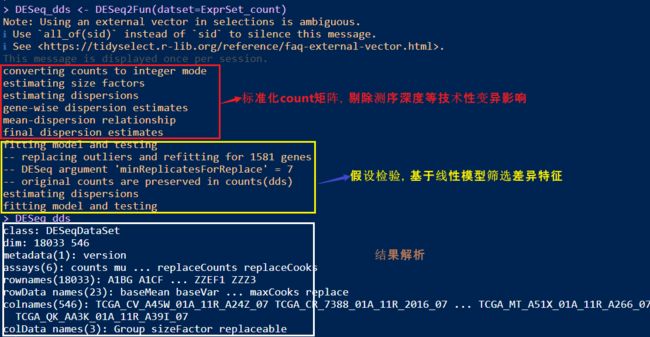
- 提取结果
DESeq_result <- results(DESeq_dds, contrast = c("Group", rev(grp)))
head(DESeq_result)
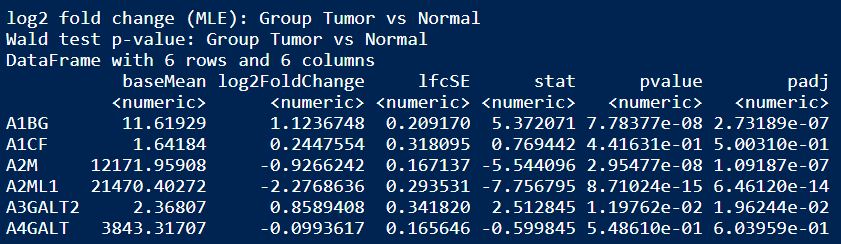
差异分组
设置差异基因过滤阈值(Foldchange+adjPval),上图设置的比较组顺序是Tumor vs Normal,所以log2foldchange > 0是Tumor组,反之则是Normal组。
DESeq_result_df <- DESeq_result %>% data.frame() %>% arrange(log2FoldChange, padj)
log2fc_threshold <- with(DESeq_result_df, mean(abs(log2FoldChange)) + 1.5 * sd(abs(log2FoldChange)))
message(paste("Threshold of log2Foldchange [Mean+1.5(SD)] is", log2fc_threshold))
pval <- 0.05
DESeq_result_df[which(DESeq_result_df$log2FoldChange >= log2fc_threshold & DESeq_result_df$padj < pval), "Enrichment"] <- grp[2]
DESeq_result_df[which(DESeq_result_df$log2FoldChange <= -log2fc_threshold & DESeq_result_df$padj < pval), "Enrichment"] <- grp[1]
DESeq_result_df[which(abs(DESeq_result_df$log2FoldChange) < log2fc_threshold | DESeq_result_df$padj >= pval), "Enrichment"] <- "Nonsignf"
table(DESeq_result_df$Enrichment)
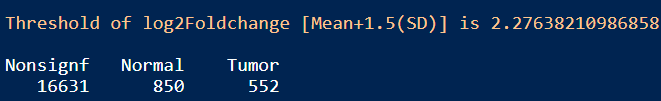
结果:差异显著的基因数目在Tumor和Normal组分别是552和850,可通过火山图展示差异基因。
Volcano plot
library(ggrepel)
VolcanoFun <- function(datset=DESeq_result_df,
genelist=c("SMR3B", "BPIFA2", "HTN1", "NOBOX", "MAGEA9B", "MAGEA10"),
group_name=grp,
group_col=grp.col,
Pval=0.05,
LogFC=round(log2fc_threshold, 2)){
# datset=DESeq_result_df
# genelist=c("SMR3B", "BPIFA2", "HTN1", "NOBOX", "MAGEA9B", "MAGEA10")
# group_name=grp
# group_col=grp.col
# Pval=0.05
# LogFC=log2fc_threshold
dat <- datset %>% rownames_to_column("FeatureID") %>%
mutate(color=factor(Enrichment,
levels = c(group_name, "Nonsignif")))
# print(table(dat$color))
dat_status <- table(dat$color)
dat_status_number <- as.numeric(dat_status)
dat_status_name <- names(dat_status)
legend_label <- c(paste0(dat_status_name[1], " (", dat_status_number[1], ")"),
paste0(dat_status_name[2], " (", dat_status_number[2], ")"),
paste0("Nonsignif", " (", dat_status_number[3], ")"))
dat.signif <- subset(dat, padj < Pval & abs(log2FoldChange) > LogFC) %>%
filter(FeatureID%in%genelist)
print(table(dat.signif$color))
group_col_new <- c(rev(group_col), "grey80")
group_name_new <- levels(dat$color)
xlabel <- paste0("log2(", paste(rev(group_name), collapse="/"), ")")
# Make a basic ggplot2 object with x-y values
pl <- ggplot(dat, aes(x=log2FoldChange, y=-log10(padj), color=color))+
geom_point(size=1, alpha=1, stroke=1)+
scale_color_manual(name=NULL,
values=group_col_new,
labels=c(legend_label, "Nonsignif"))+
xlab(xlabel) +
ylab(expression(-log[10]("adjusted p-value")))+
geom_hline(yintercept=-log10(Pval), alpha=.8, linetype=2, size=.7)+
geom_vline(xintercept=LogFC, alpha=.8, linetype=2, size=.7)+
geom_vline(xintercept=-LogFC, alpha=.8, linetype=2, size=.7)+
geom_text_repel(data = dat.signif,
aes(label = FeatureID),
size = 4,
max.overlaps = getOption("ggrepel.max.overlaps", default = 80),
segment.linetype = 1,
segment.curvature = -1e-20,
box.padding = unit(0.35, "lines"),
point.padding = unit(0.3, "lines"),
arrow = arrow(length = unit(0.005, "npc")),
color = "black", # text color
bg.color = "white", # shadow color
bg.r = 0.15)+
annotate("text", x=min(dat$log2FoldChange), y=-log10(Pval), label=Pval, size=6, color="red")+
annotate("text", x=LogFC, y=0, label=LogFC, size=6, color="red")+
annotate("text", x=-LogFC, y=0, label=-LogFC, size=6, color="red")+
scale_y_continuous(trans = "log1p")+
guides(color=guide_legend(override.aes = list(size = 3)))+
theme_bw()+
theme(axis.title = element_text(color = "black", size = 12),
axis.text = element_text(color = "black", size = 10),
text = element_text(size = 8, color = "black", family="serif"),
panel.grid = element_blank(),
#legend.position = "right",
legend.position = c(.15, .1),
legend.key.height = unit(0.6,"cm"),
legend.text = element_text(face = "bold", color = "black", size = 8),
strip.text = element_text(face = "bold", size = 14))
return(pl)
}
VolcanoFun(datset=DESeq_result_df, genelist=c("SMR3B", "BPIFA2", "HTN1", "NOBOX", "MAGEA9B", "MAGEA10"))

Notes: log2Foldchange > 0是富集在Tumor组,图上表示NOBOX, MAGEA9B, MAGEA10 是富集在Tumor组,也可以从公式log2(Tumor/Normal)看出。为了验证上述结论,使用boxplot图展示结果。
boxplot
BoxplotFun <- function(datset=ExprSet_count,
genelist=c("SMR3B", "BPIFA2", "HTN1", "NOBOX", "MAGEA9B", "MAGEA10"),
group_name=grp,
group_col=grp.col){
# datset=ExprSet_count
# genelist=c("SMR3B", "BPIFA2", "HTN1", "NOBOX", "MAGEA9B", "MAGEA10")
# group_name=grp
# group_col=grp.col
pheno <- pData(datset) %>%
rownames_to_column("SampleID") %>%
filter(Group%in%group_name) %>%
mutate(Group=factor(Group, levels = group_name)) %>%
column_to_rownames("SampleID")
print(table(pheno$Group))
edata <- data.frame(exprs(datset)) %>%
dplyr::select(rownames(pheno)) %>%
rownames_to_column("FeatureID") %>%
filter(FeatureID%in%genelist) %>%
column_to_rownames("FeatureID")
mdat <- pheno %>% dplyr::select(Group) %>%
rownames_to_column("SampleID") %>%
inner_join(t(edata) %>% data.frame() %>% rownames_to_column("SampleID"), by = "SampleID") %>%
column_to_rownames("SampleID")
plotdata <- mdat %>% tidyr::gather(key="FeatureID", value="value", -Group) %>%
mutate(Group=factor(Group, levels = group_name))
plotdata$FeatureID <- factor(plotdata$FeatureID, levels = genelist)
pl <- ggplot(plotdata, aes(x=Group, y=value, fill=Group))+
stat_boxplot(geom="errorbar", width=0.15,
position=position_dodge(0.4)) +
geom_boxplot(width=0.4,
outlier.colour="black",
outlier.shape=21,
outlier.size=.5)+
scale_fill_manual(values=group_col)+
#ggpubr::stat_compare_means(method = "wilcox.test", comparisons = group_name)+
facet_wrap(facets="FeatureID", scales="free_y", nrow=2)+
labs(x="", y="Gene Count")+
guides(fill="none")+
theme_bw()+
theme(axis.title = element_text(color="black", size=12),
axis.text.x = element_text(color="black", size=10, hjust=.5, vjust=.5, angle=60),
text = element_text(size=8, color="black", family="serif"),
panel.grid = element_blank(),
strip.text = element_text(face="bold", size=12))
return(pl)
}
BoxplotFun(datset=ExprSet_count,
genelist=c("SMR3B", "BPIFA2", "HTN1", "NOBOX", "MAGEA9B", "MAGEA10"))

Notes: 从图中可以看出,最显著富集的基因在另一组的表达值(counts数目)几乎都为0,这说明这些基因是某组独有的基因(需要进一步查看出现率判断是否是独有还是仅仅低丰度而已,因为Tumor组样本数目要远远大于Normal组),而DESeq2通过标准化因子能区分出来,而用wilcox-test检验会出现not enough observations的警告信息,因此注释掉stat_compare_means函数。

总结:表达谱矩阵是count matrix的时,可以使用DESeq2包做假设检验(差异分析)。以前做扩增子分析的时候,OTU table也是count matrix,其实也可以用该包做假设检验,但是如果转换成Relative abundance则不可。除此之外,输入参数design还可以添加协变量进行校正处理,后面有时间再写另一篇专门介绍怎么用DESeq2(limma/edgeR)校正协变量的博客吧。
limma
limma最初是用于微阵列芯片基因表达的差异分析,但后来它提供的voom函数使其可以应用于转录组等差异分析(limma-voom模型),那我们肯定好奇voom函数的原理是什么。
Voom功能
这里要着重介绍下voom函数的作用:
-
标准化counts成CPM:log2 transform;
-
计算每个基因的log2CPM的线性模型残差(residuals = observed - fitted);
-
通过平均表达值拟合残差标准差(residual standard deviation);
-
结合拟合平滑曲线获得的每个基因的权重和log2CPM值用于后续差异分析。
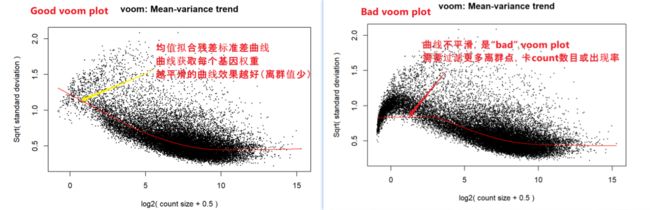
计算过程
-
构建分组矩阵;
-
构建DGEList对象;
-
计算Counts标准化因子;
-
voom标准化;
-
线性模型计算每个基因在分组的weighted least square;
-
构建比较对象;
-
计算每个基因在比较对象间的比较结果;
-
在基因的平均标准误基础上,使用经典贝叶斯算法缩小基因组间比较结果的最大最小标准误差;
-
提取最终差异结果。
library(limma)
Limma2Fun <- function(datset=ExprSet_count){
# datset=ExprSet_count
profile <- exprs(datset)
metadata <- pData(datset)
sid <- intersect(rownames(metadata), colnames(profile))
colData <- metadata %>% rownames_to_column("SampleID") %>%
dplyr::select(SampleID, Group) %>%
filter(SampleID%in%sid) %>%
mutate(Group=factor(Group, levels = grp)) %>%
column_to_rownames("SampleID")
countData <- profile %>% data.frame() %>%
dplyr::select(all_of(sid)) %>%
as.matrix()
if(!all(rownames(colData) == colnames(countData))){
stop("Order is wrong please check your data")
}
# group matrix
design <- model.matrix(~0 + Group, data = colData)
colnames(design) <- levels(colData$Group)
rownames(design) <- colnames(countData)
# DGEList object: counts-> colnames:Samples; rownames: Features
dge_obj <- edgeR::DGEList(counts = countData)
dge_obj_factors <- edgeR::calcNormFactors(dge_obj)
# voom
dat_voom <- voom(counts = dge_obj_factors,
design = design,
normalize.method = "quantile",
plot = TRUE)
# Linear model to calculate weighted least squares per gene in each group
fit <- lmFit(object = dat_voom,
design = design)
# Comparison between two groups
contr <- paste(rev(levels(colData$Group)), collapse = "-")
contr.matrix <- makeContrasts(contrasts = contr,
levels = design)
# Estimate contrast for each gene
fit.contr <- contrasts.fit(fit, contr.matrix)
# Empirical Bayes smoothing of standard errors
fit.ebay <- eBayes(fit.contr)
# DE result table
res_table <- topTable(fit.ebay, coef = contr, n=Inf, sort.by = "P")
return(res_table)
}
Limma_res <- Limma2Fun(datset=ExprSet_count)
head(Limma_res)
查看voom拟合曲线结果:曲线拟合不够平滑,说明需要设置counts或occurrence过滤阈值过滤离群的基因
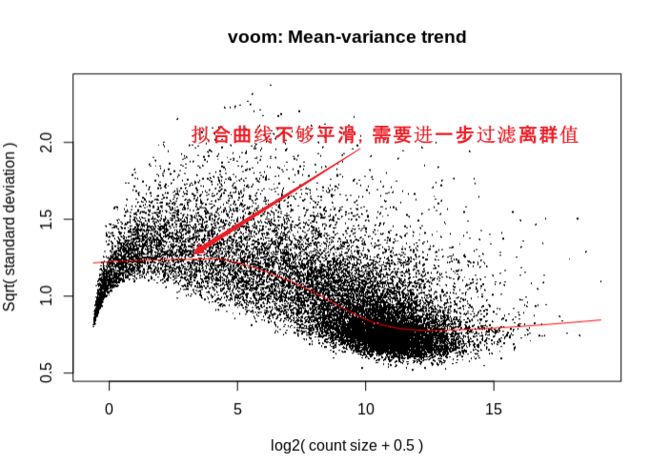
limma差异结果:

差异分组
设置差异基因过滤阈值(logFC+adj.P.Val)
Limma_res_df <- Limma_res %>% data.frame() %>% arrange(logFC, adj.P.Val)
log2fc_threshold <- with(Limma_res_df, mean(abs(logFC)) + 1.5 * sd(abs(logFC)))
message(paste("Threshold of log2Foldchange [Mean+1.5(SD)] is", log2fc_threshold))
pval <- 0.05
Limma_res_df[which(Limma_res_df$logFC >= log2fc_threshold & Limma_res_df$adj.P.Val < pval), "Enrichment"] <- grp[2]
Limma_res_df[which(Limma_res_df$logFC <= -log2fc_threshold & Limma_res_df$adj.P.Val < pval), "Enrichment"] <- grp[1]
Limma_res_df[which(abs(Limma_res_df$logFC) < log2fc_threshold | Limma_res_df$adj.P.Val >= pval), "Enrichment"] <- "Nonsignf"
table(Limma_res_df$Enrichment)
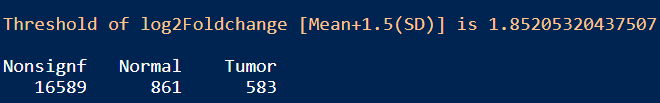
结果:差异显著的基因数目在Tumor和Normal组分别是583和861,火山图展示差异基因如上图修改代码即可。
edgeR
edgeR包也是基于线性模型做差异分析的包。
计算过程
-
先存储counts成DGEList对象;
-
过滤数据(可选择);
-
标准化counts矩阵;
-
计算离散程度(整体可变性评估estimateGLMCommonDisp;趋势模型estimateGLMTrendedDisp;比较组estimateGLMTagwiseDisp);
-
GLM 模型的likelihood ratio test进行差异分析。
library(edgeR)
EdgeR2Fun <- function(datset=ExprSet_count){
# datset=ExprSet_count
profile <- exprs(datset)
metadata <- pData(datset)
sid <- intersect(rownames(metadata), colnames(profile))
colData <- metadata %>% rownames_to_column("SampleID") %>%
dplyr::select(SampleID, Group) %>%
filter(SampleID%in%sid) %>%
mutate(Group=factor(Group, levels = grp)) %>%
column_to_rownames("SampleID")
countData <- profile %>% data.frame() %>%
dplyr::select(all_of(sid)) %>%
as.matrix()
if(!all(rownames(colData) == colnames(countData))){
stop("Order is wrong please check your data")
}
# group matrix
design <- model.matrix(~0 + Group, data = colData)
colnames(design) <- levels(colData$Group)
rownames(design) <- colnames(countData)
# Filter data
# keep <- rowSums(cpm(countData) > 100) >= 2
# countData <- countData[keep, ]
# DGEList object: counts-> colnames:Samples; rownames: Features
dge_obj <- DGEList(counts = countData)
dge_obj_factors <- calcNormFactors(dge_obj)
plotMDS(dge_obj_factors, method="bcv", col=as.numeric(colData$Group))
legend("bottomleft", as.character(unique(colData$Group)), col=1:2, pch=20)
# GLM estimates of dispersion
dge <- estimateGLMCommonDisp(dge_obj_factors, design)
dge <- estimateGLMTrendedDisp(dge, design, method="power")
# You can change method to "auto", "bin.spline", "power", "spline", "bin.loess".
# The default is "auto" which chooses "bin.spline" when > 200 tags and "power" otherwise.
dge <- estimateGLMTagwiseDisp(dge, design)
plotBCV(dge)
# GLM testing for differential expression:
fit <- glmFit(dge, design)
fit2 <- glmLRT(fit, contrast=c(-1, 1)) # Normal:-1; Tumor:1
res_table <- topTags(fit2, n=nrow(countData)) %>% data.frame()
return(res_table)
}
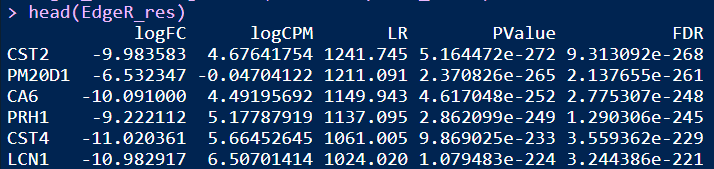
EdgeR_res <- EdgeR2Fun(datset=ExprSet_count)
head(EdgeR_res)
差异分组
设置差异基因过滤阈值(logFC+FDR)
EdgeR_res_df <- EdgeR_res %>% arrange(logFC, FDR)
log2fc_threshold <- with(EdgeR_res_df, mean(abs(logFC)) + 1.5 * sd(abs(logFC)))
message(paste("Threshold of log2Foldchange [Mean+1.5(SD)] is", log2fc_threshold))
pval <- 0.05
EdgeR_res_df[which(EdgeR_res_df$logFC >= log2fc_threshold & EdgeR_res_df$FDR < pval), "Enrichment"] <- grp[2]
EdgeR_res_df[which(EdgeR_res_df$logFC <= -log2fc_threshold & EdgeR_res_df$FDR < pval), "Enrichment"] <- grp[1]
EdgeR_res_df[which(abs(EdgeR_res_df$logFC) < log2fc_threshold | EdgeR_res_df$FDR >= pval), "Enrichment"] <- "Nonsignf"
table(EdgeR_res_df$Enrichment)
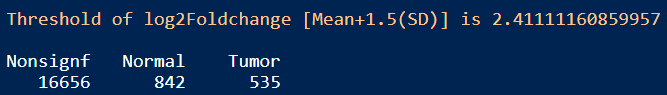
结果:差异显著的基因数目在Tumor和Normal组分别是535和842,火山图展示差异基因如上图修改代码即可。
Wilcox-rank-sum test or t-test
判断基因的表达值是否符合正态分布,是则选择t-test,否则选择wilcox-rank-sum test,log2foldchange根据scale结果的两组均值相除获取。
计算差异结果
DEA_test <- function(datset=ExprSet_count,
group_info="Group",
group_name=grp){
# datset=ExprSet
# group_info="Group"
# group_name=grp
phenotype <- pData(datset)
profile <- exprs(datset)
phenotype_cln <- phenotype %>% dplyr::select(all_of(group_info))
colnames(phenotype_cln)[which(colnames(phenotype_cln) == group_info)] <- "Group"
pheno <- phenotype_cln %>%
rownames_to_column("SampleID") %>%
filter(Group%in%group_name) %>%
mutate(Group=factor(Group, levels = group_name)) %>%
column_to_rownames("SampleID")
print(table(pheno$Group))
edata <- profile[, rownames(pheno)]
if(length(unique(pheno$Group)) != 2){
stop("Levels of Group must be 2 levels")
}
# Checking whether the two groups that where tested were normally distributed using the Shapiro-Wilk test
shapiro_res <- apply(edata, 1, function(x, y){
#dat <- data.frame(value=as.numeric(edata[1, ]), group=pheno$Group)
dat <- data.frame(value=x, group=y)
nUnique <- tapply(dat$value, dat$group, function(x){length(unique(x))})
if(nUnique[1] < 5 | nUnique[2] < 5){
res <- NA
}else{
t_res <- tapply(dat$value, dat$group, shapiro.test)
if(t_res[[1]]$p.value < 0.05){
res <- FALSE
}else{
res <- TRUE
}
}
return(res)
}, pheno$Group) %>%
data.frame() %>%
setNames("Normal") %>%
rownames_to_column("FeatureID")
shapiro_res_final <- shapiro_res %>% filter(!is.na(Normal))
Normal_prof <- edata[rownames(edata)%in%shapiro_res_final$Normal, ]
Non_Normal_prof <- edata[!rownames(edata)%in%shapiro_res_final$Normal, ]
if(nrow(Normal_prof) != 0){
# Normally distributed proteins
Welch_res <- apply(Normal_prof, 1, function(x, y){
dat <- data.frame(value=as.numeric(x), group=y)
mn_GM <- tapply(dat$value_scale, dat$group, compositions::geometricmean) %>%
data.frame() %>% setNames("value") %>%
rownames_to_column("Group")
mn_GM1 <- with(mn_GM, mn_GM[Group%in%group_name[1], "value"])
mn_GM2 <- with(mn_GM, mn_GM[Group%in%group_name[2], "value"])
Log2FC_GM <- log2(mn_GM1/mn_GM2)
# pvalue
rest <- rstatix::t_test(data = dat, value ~ group)
return(c(Log2FC_GM, mn_GM1, mn_GM2,
rest$statistic, rest$p))
}, pheno$Group) %>%
t() %>% data.frame() %>%
setNames(c("logFC", paste0("geometricmean_", group_name),
"Statistic", "P.value"))
Normal_res <- Welch_res %>%
rownames_to_column("FeatureID") %>%
arrange(desc(abs(logFC)), P.value)
}else{
Normal_res <- data.frame()
}
if(nrow(Non_Normal_prof) != 0){
# non-Normally distributed proteins
Wilcox_res <- apply(Non_Normal_prof, 1, function(x, y){
dat <- data.frame(value=as.numeric(x), group=y)
# Fold2Change
dat$value_scale <- scale(dat$value, center = TRUE, scale = TRUE)
mn_GM <- tapply(dat$value_scale, dat$group, compositions::geometricmean) %>%
data.frame() %>% setNames("value") %>%
rownames_to_column("Group")
mn_GM1 <- with(mn_GM, mn_GM[Group%in%group_name[1], "value"])
mn_GM2 <- with(mn_GM, mn_GM[Group%in%group_name[2], "value"])
Log2FC_GM <- log2(mn_GM1/mn_GM2)
# pvalue
rest <- wilcox.test(data = dat, value ~ group)
return(c(Log2FC_GM, mn_GM1, mn_GM2,
rest$statistic, rest$p.value))
}, pheno$Group) %>%
t() %>% data.frame() %>%
setNames(c("logFC", paste0("geometricmean_", group_name),
"Statistic", "P.value"))
Non_Normal_res <- Wilcox_res %>%
rownames_to_column("FeatureID") %>%
arrange(desc(abs(logFC)), P.value)
}else{
Non_Normal_res <- data.frame()
}
# Number & Block
res <- rbind(Normal_res, Non_Normal_res)
res$adj.P.Val <- p.adjust(as.numeric(res$P.value), method = "BH")
dat_status <- table(pheno$Group)
dat_status_number <- as.numeric(dat_status)
dat_status_name <- names(dat_status)
res$Block <- paste(paste(dat_status_number[1], dat_status_name[1], sep = "_"),
"vs",
paste(dat_status_number[2], dat_status_name[2], sep = "_"))
res_final <- res %>% dplyr::select(FeatureID, Block, adj.P.Val, P.value, everything()) %>%
arrange(adj.P.Val)
return(res_final)
}
Test_res <- DEA_test(datset=ExprSet_count)
head(Test_res)

差异分组
设置差异基因过滤阈值(logFC+adj.P.Va)
Test_res_df <- Test_res %>% filter(!is.nan(logFC)) %>%
filter(!is.infinite(logFC)) %>%
filter(is.finite(logFC)) %>%
arrange(logFC, adj.P.Val)
log2fc_threshold <- with(Test_res_df, mean(abs(logFC)) + 1.5 * sd(abs(logFC)))
message(paste("Threshold of log2Foldchange [Mean+1.5(SD)] is", log2fc_threshold))
pval <- 0.05
Test_res_df[which(Test_res_df$logFC >= log2fc_threshold & Test_res_df$adj.P.Va < pval), "Enrichment"] <- grp[1]
Test_res_df[which(Test_res_df$logFC <= -log2fc_threshold & Test_res_df$adj.P.Va < pval), "Enrichment"] <- grp[2]
Test_res_df[which(abs(Test_res_df$logFC) < log2fc_threshold | Test_res_df$adj.P.Va >= pval), "Enrichment"] <- "Nonsignf"
table(Test_res_df$Enrichment)
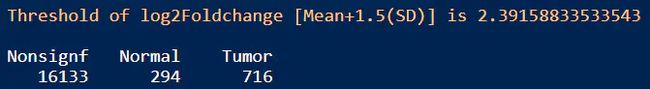
结果:差异显著的基因数目在Tumor和Normal组分别是716和294,火山图展示差异基因如上图修改代码即可。
不同方法的结果比较
虽然log2FoldChange值都是使用mean+1.5SD(均值+1.5倍方差),但是会发现四种方法的最后的log2FoldChange的阈值均不相同,这也是导致差异基因数目不同的原因之一。
保存结果
save(DESeq_result_df, Limma_res_df, EdgeR_res_df, Test_res_df, file = "DEA.RData")
差异基因数目分布
DEA_status <- data.frame(DESeq2=as.numeric(table(DESeq_result_df$Enrichment)),
Limma=as.numeric(table(Limma_res_df$Enrichment)),
EdgeR=as.numeric(table(EdgeR_res_df$Enrichment)),
T_wilcox=as.numeric(table(Test_res_df$Enrichment)))
rownames(DEA_status) <- c("Non-significant", grp)
#DT::datatable(DEA_status)
DEA_status
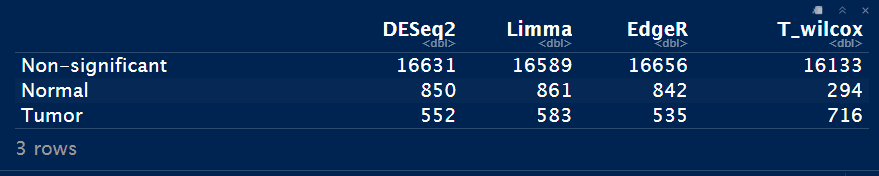
Notes: T_wilcox方法的差异基因数目是最少的。
差异基因交集Venn
Venn画图可参考R可视化:ggplot语法的Venn 图
DESeq_DEG <- DESeq_result_df %>% filter(Enrichment != "Nonsignf")
Limma_DEG <- Limma_res_df %>% filter(Enrichment != "Nonsignf")
EdgeR_DEG <- EdgeR_res_df %>% filter(Enrichment != "Nonsignf")
Test_DEG <- Test_res_df %>% filter(Enrichment != "Nonsignf")
library(ggVennDiagram)
dat_DEG <- list(
A = DESeq_DEG$FeatureID,
B = Limma_DEG$FeatureID,
C = EdgeR_DEG$FeatureID,
D = Test_DEG$FeatureID)
ggVennDiagram(dat_DEG, label_alpha = 0,
category.names = c("DESeq2", "Limma+Voom", "EdgeR", "T-test/WilcoxTest")) +
scale_fill_gradient(low = "blue", high = "yellow")+
ggtitle("Comprison of Differential Expression Approaches")
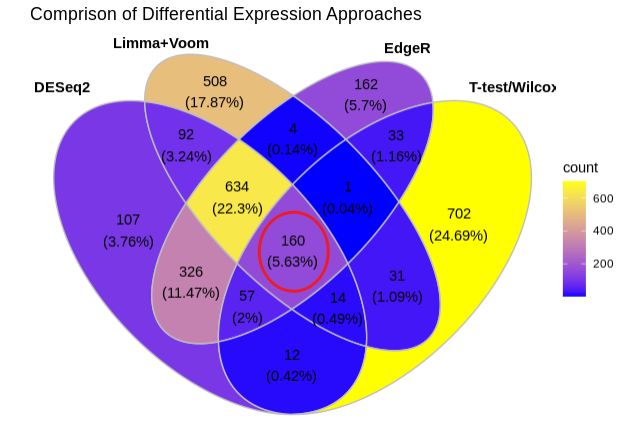
Notes:
-
四种方法重叠的差异基因数目是160个;
-
DEseq2和Limma+Voom的重叠差异基因数目:92+634+160+14;
-
DEseq2和EdgeR的重叠差异基因数目:634+326+57+160;
-
DEseq2和T-test/WilcoxTest的重叠差异基因数目:160+57+12+14;
-
DEseq2,Limma+Voom和EdgeR的重叠差异基因数目:634+160;
-
T-test/WilcoxTest和其他三种方法的重叠差异基因数目最少。
差异基因heatmap+volcano
参考3大差异分析r包:DESeq2、edgeR和limma
library(tinyarray)
dat_count <- exprs(ExprSet_count)
dat_group <- pData(ExprSet_count)
h1 <- draw_heatmap(dat_count[DESeq_DEG$FeatureID, ], factor(dat_group$Group), n_cutoff = 2)
h2 <- draw_heatmap(dat_count[Limma_DEG$FeatureID, ], factor(dat_group$Group), n_cutoff = 2)
h3 <- draw_heatmap(dat_count[EdgeR_DEG$FeatureID, ], factor(dat_group$Group), n_cutoff = 2)
h4 <- draw_heatmap(dat_count[Test_DEG$FeatureID, ], factor(dat_group$Group), n_cutoff = 2)
m2d <- function(x){
mean(abs(x)) + 1.5 * sd(abs(x))
}
v1 <- draw_volcano(DESeq_result_df %>% column_to_rownames("FeatureID"), pkg = 1, logFC_cutoff = m2d(DESeq_result_df$log2FoldChange))
v2 <- draw_volcano(Limma_res_df %>% column_to_rownames("FeatureID"), pkg = 3, logFC_cutoff = m2d(Limma_res_df$logFC))
v3 <- draw_volcano(EdgeR_res_df %>% column_to_rownames("FeatureID"), pkg = 2, logFC_cutoff = m2d(EdgeR_res_df$logFC))
# convert Test_res_df format into DESeq_result_df format
Test_res_df_DESeq <- Test_res_df %>% dplyr::rename(baseMean=geometricmean_Tumor,
log2FoldChange=logFC,
lfcSE=geometricmean_Normal,
stat=Statistic,
pvalue=P.value,
padj=adj.P.Val) %>%
dplyr::select(FeatureID, baseMean, log2FoldChange, lfcSE, stat, pvalue, padj, Enrichment)
v4 <- draw_volcano(Test_res_df_DESeq %>% column_to_rownames("FeatureID"), pkg = 1, logFC_cutoff = m2d(Test_res_df_DESeq$log2FoldChange))+
xlab("T-test/WilcoxTest")
(h1 + h2 + h3 + h4 + v1 + v2 + v3 + v4) +
plot_layout(guides = "collect", nrow = 2) & theme(legend.position = "none")
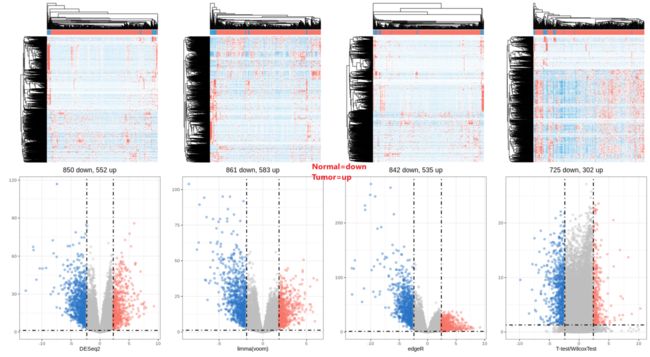
Notes: 火山图和热图也表明前三种差异检验方法差异基因趋势相对一致,而最后一种与前面三种均存在差异,这提示我们用counts矩阵在t-test/wilcox-rank-sum test中做假设检验时候要非常小心注意(也说明测序深度对假设检验结果影响较大)。
共有差异基因venn
综上,我们提取前三种方法(DEseq2,Limma+Voom和EdgeR)的重叠差异基因做后续分析,鉴别三种方法的优劣。
DESeq_DEG <- DESeq_result_df %>% filter(Enrichment != "Nonsignf")
Limma_DEG <- Limma_res_df %>% filter(Enrichment != "Nonsignf")
EdgeR_DEG <- EdgeR_res_df %>% filter(Enrichment != "Nonsignf")
ExtractDEG <- function(datDEG=DESeq_DEG,
tag=grp[1]){
tmp <- datDEG %>% filter(Enrichment == tag)
res <- tmp$FeatureID
return(res)
}
DESeq_DEG_normal <- ExtractDEG(datDEG=DESeq_DEG, tag=grp[1])
DESeq_DEG_tumor <- ExtractDEG(datDEG=DESeq_DEG, tag=grp[2])
Limma_DEG_normal <- ExtractDEG(datDEG=Limma_DEG, tag=grp[1])
Limma_DEG_tumor <- ExtractDEG(datDEG=Limma_DEG, tag=grp[2])
EdgeR_DEG_normal <- ExtractDEG(datDEG=EdgeR_DEG, tag=grp[1])
EdgeR_DEG_tumor <- ExtractDEG(datDEG=EdgeR_DEG, tag=grp[2])
DEG_normal <- intersect(DESeq_DEG_normal, intersect(Limma_DEG_normal, EdgeR_DEG_normal))
DEG_tumor <- intersect(DESeq_DEG_tumor, intersect(Limma_DEG_tumor, EdgeR_DEG_tumor))
draw_venn(list(DESeq2=DESeq_DEG_normal,
Limma=Limma_DEG_normal,
EdgeR=EdgeR_DEG_normal),
"DEG in Normal") +
draw_venn(list(DESeq2=DESeq_DEG_tumor,
Limma=Limma_DEG_tumor,
EdgeR=EdgeR_DEG_tumor),
"DEG in Tumor")
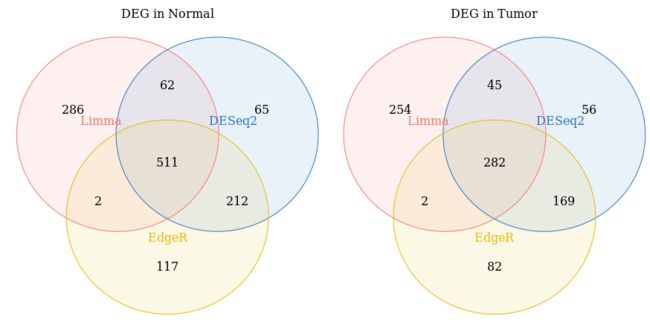
Notes: 三种方法富集在Normal和Tumor组的差异基因还是有区别的。接下来我们用相关性图展示三种方法的差异基因的相关性。
差异基因Log2Foldchange相关性分析
差异基因的Log2Foldchange相关性分析 (参考文章转录组分析5——差异表达分析)
library(corrplot)
All_DEG <- unique(c(DESeq_DEG$FeatureID, Limma_DEG$FeatureID, EdgeR_DEG$FeatureID))
length(All_DEG)
All_DEG_lg2FC <- data.frame(DESeq2=DESeq_result_df[DESeq_result_df$FeatureID%in%All_DEG, 3],
Limma=Limma_res_df[Limma_res_df$FeatureID%in%All_DEG, 2],
EdgeR=EdgeR_res_df[EdgeR_res_df$FeatureID%in%All_DEG, 2]) %>%
na.omit()
All_DEG_lg2FC_cor <- cor(All_DEG_lg2FC)
All_DEG_lg2FC_cor
corrplot(All_DEG_lg2FC_cor)
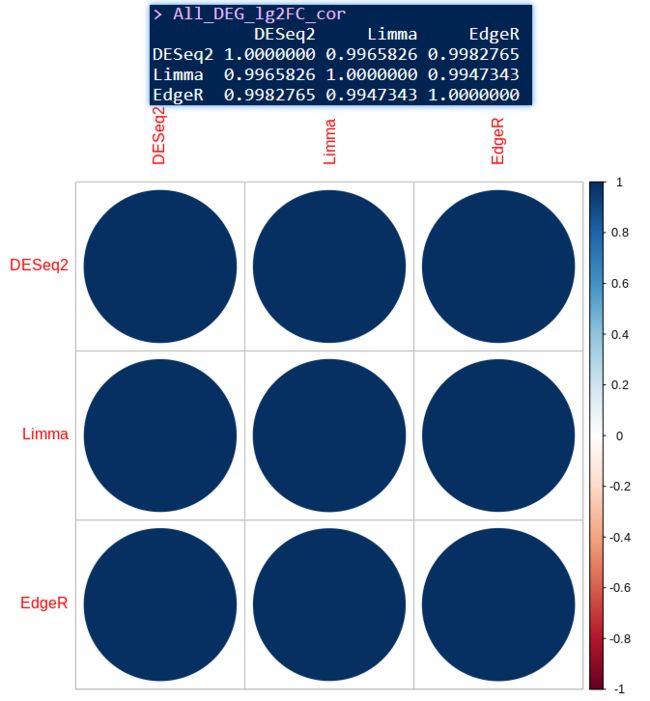
共有差异基因总结
三种方法的差异基因的PCA+heatmap+Venn(参考3大差异分析r包:DESeq2、edgeR和limma)
DEG_normal <- intersect(DESeq_DEG_normal, intersect(Limma_DEG_normal, EdgeR_DEG_normal))
DEG_tumor <- intersect(DESeq_DEG_tumor, intersect(Limma_DEG_tumor, EdgeR_DEG_tumor))
dat_count_log <- log2(cpm(dat_count)+1)
DEG_heatmap <- draw_heatmap(dat_count_log[c(DEG_normal, DEG_tumor), ], factor(dat_group$Group), n_cutoff = 2)
DEG_pca <- draw_pca(dat_count_log[c(DEG_normal, DEG_tumor), ], factor(dat_group$Group))
DEG_Normal_venn <- draw_venn(list(DESeq2=DESeq_DEG_normal,
Limma=Limma_DEG_normal,
EdgeR=EdgeR_DEG_normal),
"DEG in Normal")
DEG_Tumor_venn <- draw_venn(list(DESeq2=DESeq_DEG_tumor,
Limma=Limma_DEG_tumor,
EdgeR=EdgeR_DEG_tumor),
"DEG in Tumor")
(DEG_pca + DEG_heatmap + DEG_Normal_venn + DEG_Tumor_venn) +
plot_layout(nrow = 2, guides = "collect")

总结
-
有文章说edgeR分析速度快,但是从这次分析看,它反而是最慢的,另外edgeR的假阳性太高;
-
DESeq2在计算标准化因子时耗时太久,但它的标准化因子相对来说最合理;
-
三种方法得到的差异基因不是完全重叠的,但再提取它们所有的差异基因log2Foldchange值做相关性分析,发现相似度极高,也就是说明三种方法差异不是很大;
-
最后大家选择适合自己的方法做差异分析即可。
systemic information
sessionInfo()
R version 4.0.2 (2020-06-22)
Platform: x86_64-conda_cos6-linux-gnu (64-bit)
Running under: CentOS Linux 8 (Core)
Matrix products: default
BLAS/LAPACK: /disk/share/anaconda3/lib/libopenblasp-r0.3.10.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8 LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats4 parallel stats graphics grDevices utils datasets methods base
other attached packages:
[1] corrplot_0.84 tinyarray_2.2.7 ggVennDiagram_0.5.0 edgeR_3.32.1
[5] ggrepel_0.9.1.9999 DESeq2_1.30.1 SummarizedExperiment_1.20.0 MatrixGenerics_1.2.1
[9] matrixStats_0.59.0 GenomicRanges_1.42.0 GenomeInfoDb_1.26.4 IRanges_2.24.1
[13] S4Vectors_0.28.1 convert_1.64.0 marray_1.68.0 limma_3.46.0
[17] Biobase_2.50.0 BiocGenerics_0.36.0 cowplot_1.1.0 patchwork_1.0.1
[21] ggplot2_3.3.5 data.table_1.14.0 tibble_3.1.5 dplyr_1.0.7
loaded via a namespace (and not attached):
[1] utf8_1.2.1 reticulate_1.18 tidyselect_1.1.1 RSQLite_2.2.5 AnnotationDbi_1.52.0
[6] htmlwidgets_1.5.3 FactoMineR_2.4 grid_4.0.2 BiocParallel_1.24.1 scatterpie_0.1.5
[11] munsell_0.5.0 units_0.7-2 DT_0.18 umap_0.2.7.0 withr_2.4.2
[16] colorspace_2.0-2 GOSemSim_2.16.1 knitr_1.33 leaps_3.1 rstudioapi_0.13
[21] robustbase_0.93-6 bayesm_3.1-4 ggsignif_0.6.0 DOSE_3.16.0 labeling_0.4.2
[26] GenomeInfoDbData_1.2.4 KMsurv_0.1-5 polyclip_1.10-0 bit64_4.0.5 farver_2.1.0
[31] pheatmap_1.0.12 downloader_0.4 vctrs_0.3.8 generics_0.1.0 lambda.r_1.2.4
[36] xfun_0.24 R6_2.5.0 graphlayouts_0.7.1 locfit_1.5-9.4 gridGraphics_0.5-1
[41] bitops_1.0-7 cachem_1.0.5 fgsea_1.16.0 DelayedArray_0.16.3 assertthat_0.2.1
[46] scales_1.1.1 ggraph_2.0.4 enrichplot_1.10.1 gtable_0.3.0 tidygraph_1.2.0
[51] rlang_0.4.11 genefilter_1.72.0 scatterplot3d_0.3-41 splines_4.0.2 rstatix_0.7.0
[56] broom_0.7.9 BiocManager_1.30.16 yaml_2.2.1 reshape2_1.4.4 abind_1.4-5
[61] crosstalk_1.1.1 backports_1.2.1 qvalue_2.22.0 clusterProfiler_3.18.0 tensorA_0.36.2
[66] tools_4.0.2 ggplotify_0.0.5 ellipsis_0.3.2 jquerylib_0.1.4 RColorBrewer_1.1-2
[71] proxy_0.4-26 Rcpp_1.0.7 plyr_1.8.6 zlibbioc_1.36.0 classInt_0.4-3
[76] purrr_0.3.4 RCurl_1.98-1.3 ggpubr_0.4.0 openssl_1.4.4 viridis_0.6.1
[81] zoo_1.8-8 cluster_2.1.0 haven_2.3.1 factoextra_1.0.7 tinytex_0.32
[86] magrittr_2.0.1 RSpectra_0.16-0 futile.options_1.0.1 DO.db_2.9 openxlsx_4.2.3
[91] survminer_0.4.9 hms_1.1.0 evaluate_0.14 xtable_1.8-4 XML_3.99-0.6
[96] VennDiagram_1.6.20 rio_0.5.16 readxl_1.3.1 gridExtra_2.3 compiler_4.0.2
[101] KernSmooth_2.23-18 crayon_1.4.1 shadowtext_0.0.7 htmltools_0.5.1.1 tidyr_1.1.4
[106] geneplotter_1.66.0 DBI_1.1.1 tweenr_1.0.1 formatR_1.11 MASS_7.3-54
[111] sf_0.9-5 compositions_2.0-2 Matrix_1.3-4 car_3.0-10 cli_3.0.1
[116] igraph_1.2.6 km.ci_0.5-2 forcats_0.5.0 pkgconfig_2.0.3 flashClust_1.01-2
[121] rvcheck_0.1.8 foreign_0.8-81 annotate_1.68.0 bslib_0.2.5.1 XVector_0.30.0
[126] stringr_1.4.0 digest_0.6.27 rmarkdown_2.9 cellranger_1.1.0 fastmatch_1.1-0
[131] survMisc_0.5.5 curl_4.3.2 lifecycle_1.0.0 jsonlite_1.7.2 carData_3.0-4
[136] futile.logger_1.4.3 viridisLite_0.4.0 askpass_1.1 fansi_0.5.0 pillar_1.6.4
[141] lattice_0.20-44 fastmap_1.1.0 httr_1.4.2 DEoptimR_1.0-8 survival_3.2-11
[146] GO.db_3.12.1 glue_1.4.2 zip_2.1.1 bit_4.0.4 ggforce_0.3.2
[151] class_7.3-19 stringi_1.4.6 sass_0.4.0 blob_1.2.1 org.Hs.eg.db_3.12.0
[156] memoise_2.0.0 e1071_1.7-7
Reference
-
RPKM、FPKM、TPM详解
-
rpkm-fpkm-and-tpm-clearly-explained
-
DESeq2
-
DESeq2 DFrame error
-
Differential Expression with Limma-Voom
-
RNA Sequence Analysis in R: edgeR