零基础入门转录组分析——第五章(表达定量)
零基础入门转录组分析——第五章(表达定量)
目录
- 零基础入门转录组分析——第五章(表达定量)
-
- 1. 序列比对结果的复查
- 2. 表达定量
- 3. 提取有效信息
- 4. 合并多个样本定量结果
- 5. 进一步修改基因表达矩阵
(我这里使用的虚拟机是vmwarewokstation,版本16.0.0, linux系统是ubantu64位,版本20.04.3)
上一章我们做了序列比对,并对原始的XXX.sam文件进行压缩,获得XXX.bam文件。
本实验选用的是模式生物——C57BL/6J小鼠。
实验分组:药物处理组,对照组,每组6只鼠。
软件:subread中的featureCounts(如果没有安装,可以参考之前章节[零基础入门转录组分析——第一章(软件的安装)](https://blog.csdn.net/weixin_49878699/article/details/128787068))
1. 序列比对结果的复查
在前三章中,分别做了原始数据的准备和质控,质量过滤以及序列比对。
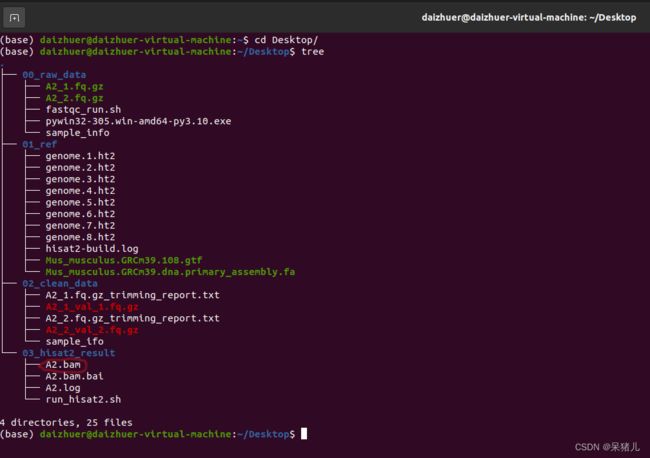
首先来看一下现有的文件和之前的输出结果如下图所示:(质控的结果我给删了,有点占地方)
- 00_raw_data文件夹中是我们的原始数据和样本信息表,但是有了clean_data这些就用不到了。
- 01_ref文件夹中有参考基因组(XXX.fa)和注释文件(XXX.gtf)以及上一章我们构建好的参考基因组索引文件(XXX.ht2)
- 02_clean_data文件夹中有之前做完质量过滤的数据(又叫做clean data)。
- 03_hisat2_result文件夹中有上一章做完的序列比对结果文件:A2.bam和A2.log
在这一章中我们会用到的文件有:
01_ref文件夹中的参考基因组注释文件(XXX.gtf)
03_hisat2_result文件夹中的序列比对结果:A2.bam文件
会用到的软件是:subread中的featureCounts,可通过指令:which featureCounts查看是否安装成功。如下图,表示安装成功。

通过指令:mkdir 04_quantify_result创建一个名为04_quantify_result的文件夹,可以后续用来存放结果文件
2. 表达定量
(1)检查00_ref文件夹中是否有XXX.gtf文件
(2)检查03_hisat2_result文件夹中是否有XXX.bam文件
(3)which featureCounts指令检查是否有featureCounts软件
如果上述三个都满足,接下来就可以开始定量了,如果任何一个不满足,可以翻看专栏下前几章内容
首先介绍几个我们会用到的featureCounts软件的常见参数:
-
-T 表示线程数,线程数越多,运行速度越快,可以通过
htop指令查看,如下图,我是四个处理器,所以最多只能设置四个
-
-a 输入的参考基因组注释文件路径
-
-p 是在双端测序的情况下使用,会统计fragment而不是read
-
-countReadPairs 计数的是(fragments)而不是read,此选项仅适用于双端测序read
-
-g 提供参考基因组注释文件中的一个id(这里我不太理解,但一般选默认gene_id即可)
-
**-t ** 指定定量的类型,默认是exon(外显子),这就是说当read落到外显子上才会被统计
-
-M 多重read会被统计
-
-O 允许多重比对,就是说当一个read比对到多个外显子时,这条read会被统计多次
-
-o 定量结果输出目录及文件名
其他的参数可以通过featureCounts -help查看,在此不过多描述!
表达定量:通过指令featureCounts -a /home/daizhuer/Desktop/01_ref/Mus_musculus.GRCm39.108.gtf -T 3 -p --countReadPairs -g gene_id -t exon -M -O -o /home/daizhuer/Desktop/04_quantify/A2 /home/daizhuer/Desktop/03_hisat2_result/A2.bam
如下图所示,表明featureCounts正常运行,并且图中显示有90.6%的reads定量到了基因的外显子上

我们可以看到04_quantify文件夹下生成两个文件,一个是定量的结果A2,另一个是表达定量输出的结果报告A2.summary

如果想查看定量结果报告,可以通过less -S A2.summary查看报告,就可以看到那些没有分配上的reads是出于什么原因,这每一行在此不详述了,可以自行搜索昂。

除了这个结果报告,最重要的就是定量结果:A2文件,同样可以通过less -S A2查看,如下图:
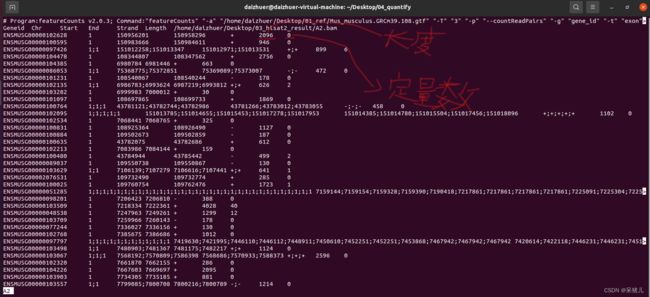
- Geneid 是基因id(重点关注)
- Chr 是该基因位于哪条染色体上
- Start 表示该基因起始位置
- End 表示该基因结束位置
- Strand 我不太懂是啥,不管!!!
- Length 基因长度,后面计算RPKM和TPM会用到(重点关注)
- 最后一列就是定量的结果(就是定量到该基因上的reads有多少条)(重点关注)
除了上述我标注的重点关注的地方,其他几个用不到,可以不关注!
3. 提取有效信息
前面我们看到A2文件中一共有7列,实际能用到的也就3列,而且这仅仅是A2一个样本的定量信息,多个样本就有多个这样的文件,所以要对文件进行处理。
通过指令cat A2 | cut -f 1,7 > A2.count可以将A2文件中的第一列和第7列切出来,并输出到A2.count文件中
通过less -S A2.count指令可以查看结果,如下图所示:

第一列是基因id,第二列就是定量到该基因上reads的数量(也就是所谓的基因表达量)。
4. 合并多个样本定量结果
通过cut指令我们得到多个修改后样本的定量结果(XXX.count),每个XXX.count都跟上图长得一样,有一列基因id,有一列定量结果,接下来要将多个样本的定量结果合并成矩阵。
我以两个样本数据作为演示(A3是复制的A2,只不过名字改成A3了)
首先我们看到当前文件夹目录下有两个.count文件

通过paste A2.count A3.count > hebing指令将两个count文件合并并输出为一个名为hebing的文件,通过less -S hebing指令可以查看结果,如下图所示:

前两列是A2.count中的内容,后两列是A3.count中的内容,多个样本可以效仿刚才的指令合并到一起。
但是实际中第一列的信息其实都一摸一样的,这就意味着我们只需要第一列的基因id和每个样本的基因表达量,那么就要提取有用的列!!!
通过指令cat hebing | cut -f 1,2,4 > raw_count就能提取hebing文件中1,2,4列,并输出结果到一个名为raw_count文件中,通过less -S raw_count命令查看,结果如下图所示:
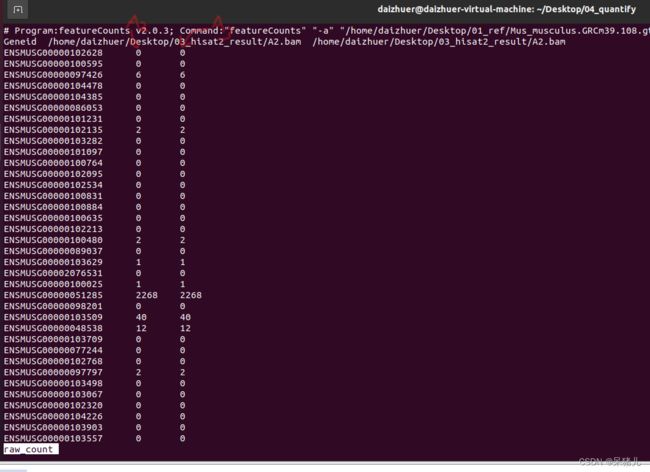
第一列是基因id,第二列是A2的基因表达量,第三列是A3的基因表达量。(如果是多个样本,可以效仿上述的命令,一定要理解才能灵活使用!!!)
5. 进一步修改基因表达矩阵
切换到linux的文件管理器下,打开04_quantify文件夹,将我们修改后的raw_count文件直接拖到宿主中。

在宿主机中找到刚才拖拽过来的raw_count文件,只接用记事本打开,修改成如下图所示的样子,就可以保存退出。

如果想用批量处理的方式自动操作,可以参考前几章的内容,在此不再反复重复了!
注:featureCounts定量结果是原始数据也就是常说的raw_count,后续还需要经过TPM和FPKM换算对数据进行标准化。
以上就是表达定量的所有过程,如果有什么需要补充或不懂的地方,大家可以私聊我或者在下方评论。
如果觉得本教程对你有所帮助,多多点赞加关注昂,下期再见!!!
同时浅述一下后续会更新的内容:(后续将会在Rstudio下进行)
- 差异表达分析和原始数据标准化(TPM和FPKM)
- 可视化之ggplot2绘制火山图
- 可视化之pheatmap绘制热图
- GO和KEGG富集分析及ggplot2绘制柱状图