lammps学习(三)聚烯烃反应力场(reaxff)热分解
目录
一、模型建立
二、in文件编写
三、问题解决
四、Conculusion
五、Reference
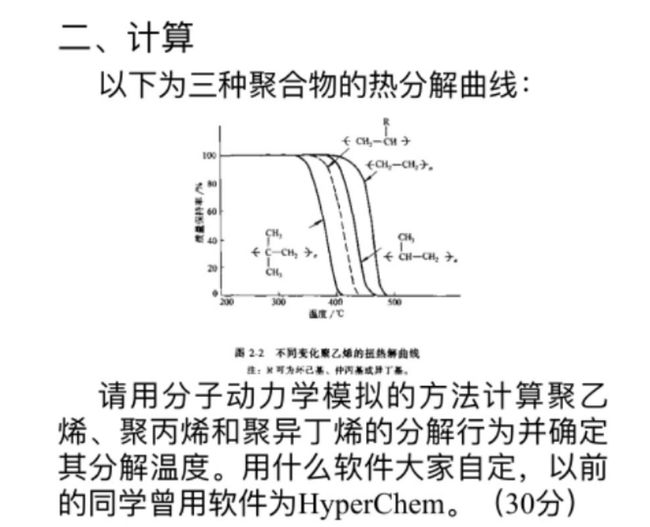
事情是这样的,师妹问了我这样一道期末试题:
我一看,这还不简单,反应力场下升温看下什么时候开始断键不就行了。说干就干,结果就做上头了。
一、模型建立
首先使用EMC生成聚合度聚60的乙烯的data文件。
由于reaxff反应力场的pair_style使用的是charge类型,只需要原子就行了,我们将data文件中和键相关的内容全部删掉。最后形式如下:
# LAMMPS data file written by OVITO Basic 3.7.5
482 atoms
2 atom types
0.0 50.0 xlo xhi
-5.0 45.0 ylo yhi
-20.0 30.0 zlo zhi
Masses
1 12.01115
2 1.00797
Atoms # charge
431 1 -0.106 42.5249956364 4.4175630658 -13.0040159035
305 1 -0.106 38.4418380355 20.7698203884 4.7169021698
416 1 -0.106 43.9936957128 9.6328217355 -11.995574631
427 2 0.053 45.1830862726 5.7435024632 -12.5229504495
434 1 -0.106 42.8528550429 4.1388712775 -11.5748594535
417 2 0.053 43.1891122967 9.7365092725 -12.6737123706
......对于这种方法生成的初始模型lammps可能无法识别,这是将data文件导入ovito以charge的形式重新输出一遍就可以了。
之后将建立好的初始模型进行一段MD得到稳定结构用于后续计算,这里我直接复制的之前写的代码。
二、in文件编写
这里我直接用的lammps自带的CHO反应力场的例子为基础修改的:
# REAX potential for CHO system
# .....
units real
atom_style charge
read_data PE.data
pair_style reax/c NULL
pair_coeff * * ffield.reax.cho C H
neighbor 2 bin
neigh_modify every 10 delay 0 check no
fix 1 all nve
fix 3 all temp/berendsen 300 1200 50.0
fix 2 all qeq/reax 1 0.0 10.0 1e-6 reax/c
fix 4 all reax/c/species 1 100 100 species.out element C H
thermo 100
thermo_style custom step temp pe ke etotal
timestep 0.25
dump 1 all custom 100 PE.xyz type x y z #每次输出升高1K
run 150000其中fix 4可以用来输出热分解产生的各种碎片的数量。运行后得到的轨迹动画如下:
然而并没有发生分解的现象。
三、问题解决
查阅相关文献和后了解到:
热分解反应是一个稀有事件,在MD体系中较低温度下有限的时间内很难观测到显著的分解现象。
因此,要解决不能观测到分解的问题,有以下两种做法:
- 增加模拟时长。
- 提高模拟温度。
这里我们选择第二种方法,上面in文件中的温度控制进行修改:
#fix 3 all temp/berendsen 300 1200 50.0
--->fix 3 all temp/berendsen 3000 3000 50.0可以看到聚乙烯发生无规断裂逐渐生成了各种小分子物质。
species.out文件内容如下:
......
# Timestep No_Moles No_Specs C20H41 C2H4 C10H19 C2H5 C4H5 H2 H CH3 C3H6 C4H8 C2H3 CH2 C2H2 C7H12 C3H5
149800 64 15 1 36 1 1 1 5 1 3 5 2 3 1 1 2 1
# Timestep No_Moles No_Specs C20H41 C2H4 C10H19 C2H5 C4H5 H2 H CH3 C3H6 C4H8 C2H3 CH2 C2H2 C7H12 C3H5
149900 64 15 1 36 1 1 1 5 1 3 5 2 3 1 1 2 1
# Timestep No_Moles No_Specs C20H41 C2H4 C10H19 C2H5 C4H5 H2 H CH3 C3H6 C4H8 C2H3 CH2 C2H2 C7H12 C3H5
150000 64 15 1 36 1 1 1 5 1 3 5 2 3 1 1 2 1
ReaxFF MD 模拟中提高反应温度仅影响反应速率,对反应机制影响较小,因此分析3000k时的结果即可研究聚烯烃分解行为。
- 在改温度下模拟37.5ps聚乙烯还未能完全分解,存在部分链长为20和10的链段。
- 共裂解生成了64个、15种链段。其中最主要的分解产物为乙烯。
- 同样的方法更换read的data文件可以研究聚丙烯和聚异丁烯的分解行为。
四、Conculusion
结果到最后似乎还是没有解决最开始关于分解温度问题。
对此我的回答是:可以算但是没必要。一方面分子动力学模拟,基于随机碰撞分解 ,计算的结果并不精确。另一方面要在较低温度下观测分解现象需要消耗大量模拟时间。因此现有的文献使用分子动力学对热分解的研究大多是设置在远高于物质分解温度的状态下。
如果有误的话欢迎各位大佬指出!
五、Reference
[1]贺兴处, 陈德珍, 梅振飞,等. CaO催化PE热解及H2O对催化过程影响的ReaxFF MD研究与机理分析[J]. 化工学报, 2021.
[2] Ramin L , Assadi M , Sahajwalla V . High-Density Polyethylene Degradation into Low Molecular Weight Gases at 1823 K: An Atomistic Simulation[J]. arXiv e-prints, 2022.
[3] LAMMPS从研一到延毕:Reax反应力场下的分子动力学模拟 LAMMPS从研一到延毕:Reax反应力场下的分子动力学模拟 - 知乎