没有涉及过microRNA的实验,所以一直没写过相关的帖子,这个星期开始,更新一下microRNA的帖子。
TCGA中的micoRNA,他的ID是这个样子的,

看起来很不和谐,但是,这种ID是可以区分出3p和5p的miRNA的。他的转换文件在这里。
http://www.mirbase.org/
以MIMAT0000062为例
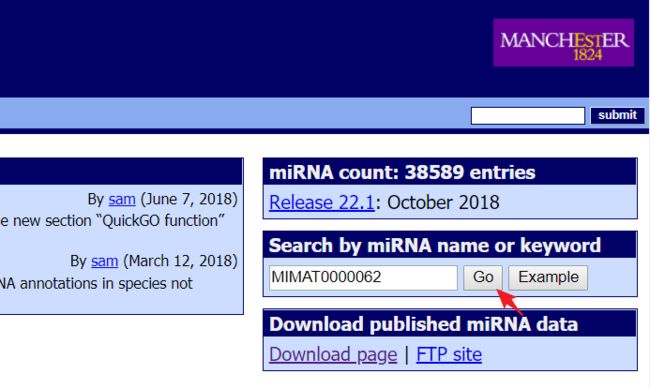
输入一个名字后点击
GO就进入以下界面:
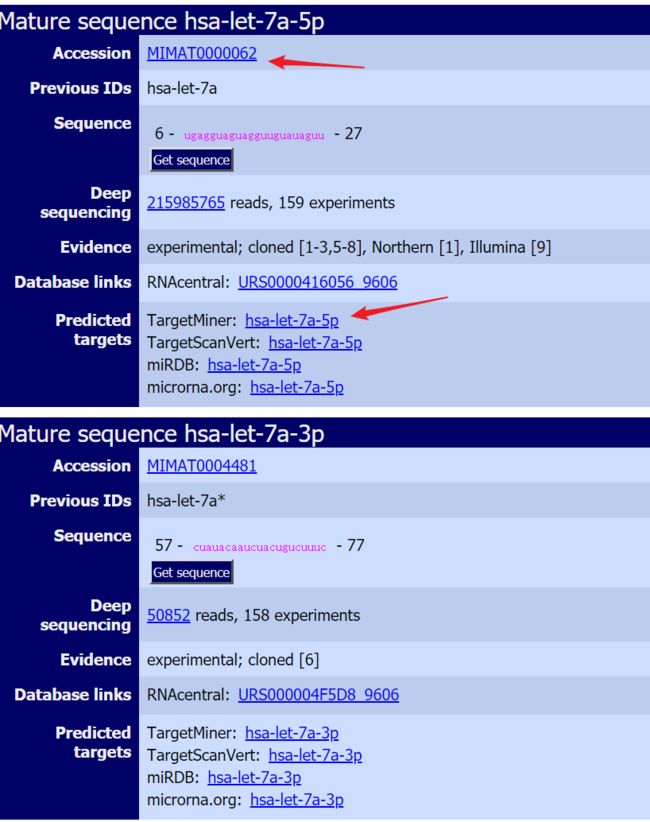
在这里,我们可以看到这个miRNA的全称是
hsa-let-7a-5p,他还有个对应的3p miRNA。
如果想要获取所有人类的miRNA转换关系呢?
主页面有个Download
里面有个fasta文件可以下载
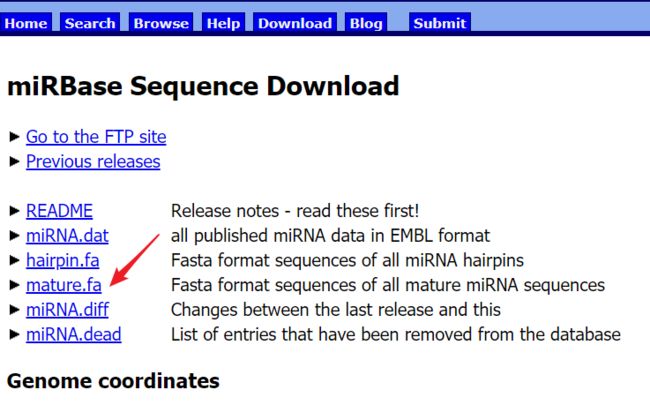
下载完了之后打开Rstudio,新建项目,把fasta文件放入工作目录对应的文件夹
解压后就是
mature.fa
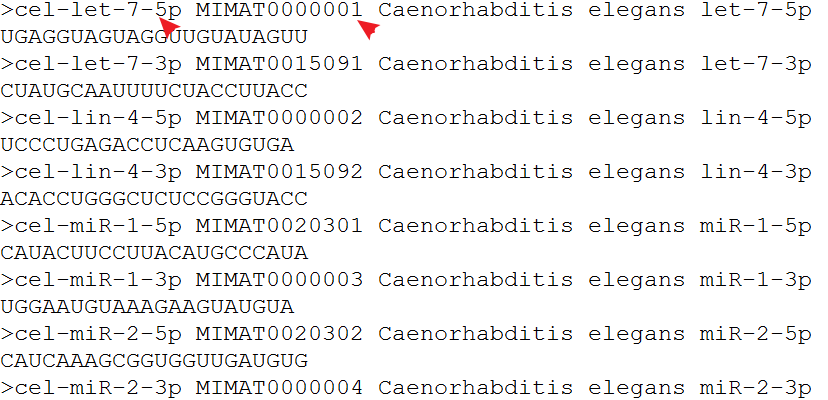
读取fasta文件,可以用Biostrings这个R包。
用readDNAStringSet这个函数读取,用names函数获取注释
library("Biostrings")
fastaFile <- readDNAStringSet("mature.fa")
rdn = names(fastaFile)
现在rdn是这个样子的
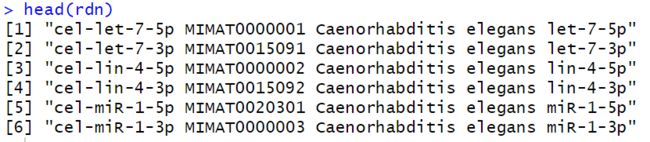
我们只要把它里面的对应关系提取出来即可,首先把它转换为数据框,就变成一列,然后用空格来裂解,选取就可以了,最终获取的是全部物种的,可以把人类的筛选出来。
dd1 <- data.frame(rdn) %>%
separate(rdn,into = c("miRNA_name","miRNA_ID","species"),sep = " ") %>%
filter(species=="Homo")
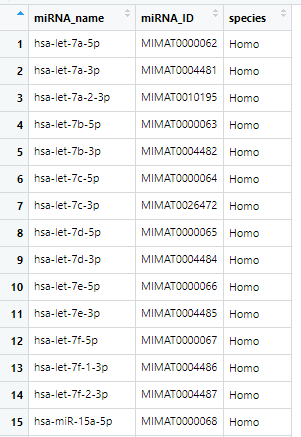
总共2656行。这样对应关系就找到了,通过限定species还可以获取其他物种的,在这里没有意义,因为TCGA是人类的样本。
还有什么方法呢,到现在我还没用过洲更和小洁介绍过的readLines,这个函数可以一行行读取文件,读取的元素就是向量的一部分
rdn <- readLines("mature.fa")
速度是又快,结果也很好

这么规则的数据,提取出想要的元素,也很简单呢
dd2 <- data.frame(rdn) %>%
## 选取 ">"
filter(grepl(">",rdn)) %>%
## 去掉 ">"
separate(rdn,into = c("drop","rdn"),sep = ">") %>%
## 分栏
separate(rdn,into = c("miRNA_name","miRNA_ID","species"),sep = " ") %>%
## 选择目标栏
select(miRNA_name,miRNA_ID,species) %>%
## 选择人类
filter(species=="Homo")
得到的结果肯定是一抹一样
这样TCGA的miRNA也可以转换了。
现在限制我们的是,没有数据嘛,那明天见。