文章题目
Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown
文章地址
https://www.nature.com/articles/nprot.2016.095.pdf
序章
- 这篇文章很牛
- STAR的比对速度是Tophat的50倍,HISAT更是STAR的1.2倍。
- STAR消耗的内存约31G(人类基因组),HISAT大约4.3G。
- StringTie的组装速度是Cufflinks的25倍,但是内存消耗却不到其一半。
- Ballgown在差异分析方面比Cuffdiff更高的特异性及准确性,且时间消耗不到Cuffdiff的千分之一。
- 简单科普
- HISAT (http://ccb.jhu.edu/software/hisat/index.shtml)利用大量FM索引,以覆盖整个基因组,能够将RNA-Seq的读取与基因组进行快速比对。它取代Bowtie/TopHat程序。
- StringTie (http://ccb.jhu.edu/software/stringtie/) 能够应用流神经网络算法和可选的de novo组装进行转录本组装并预计表达水平。与Cufflinks等程序相比,在分析模拟和真实的数据集时,StringTie实现了更完整、更准确的基因重建,并更好地预测了表达水平。
- Ballgown (https://github.com/alyssafrazee/ballgown)是基因差异表达分析的工具,能利用RNA-Seq实验的数据(StringTie, RSEM, Cufflinks的结果),预测基因、转录本的差异表达。值得注意的是,Ballgown并没有不能很好地检测差异外显子,而 DEXseq、rMATS和MISO可以很好解决该问题。
1. 环境配置
- 数据下载
# 下载数据
axel ftp://ftp.ccb.jhu.edu/pub/RNAseq_protocol/chrX_data.tar.gz
# 解压
tar xvzf chrX_data.tar.gz

- 软件安装
- HISAT2 software (http://ccb.jhu.edu/software/hisat2 or http://github.com/ infphilo/hisat2, version 2.0.1 or later)
- StringTie software (http://ccb.jhu.edu/software/stringtie or https://github. com/gpertea/stringtie , version 1.2.2 or later)
- SAMtools (http://samtools.sourceforge.net, version 0.1.19 or later)
- R (https://www.r-project.org, version 3.2.2 or later)
conda install hisat2
conda install stringtie
conda install samtools
- R 自己安装(相信你可以的!)
- conda 安装见转录组入门一(mac版):配置软件安装
2. 分析流程

这个protocol首先从原始RAN-seq数据入手,输出数据包括基因list,转录本,及每个样本的表达量,能够表现差异表达基因的表格并完成显著性的计算。
使用HISAT将读段匹配到参考基因组上,使用者可以提供注释文件,但HISAT依旧会检测注释文件没有列出来的剪切位点
比对上的reads将会被呈递给StringTie进行转录本组装,StringTie单独的对每个样本进行组装,在组装的过程中顺带估算每个基因及isoform的表达水平
所有的转录本都被呈递给StringTie的merge函数进行merge,这一步是必须的,因为有些样本的转录本可能仅仅被部分reads覆盖,无法被第二步的StringTie组装出来。merge步骤可以创建出所有样本里面都有的转录本,方便下一步的对比
merge的数据再一次被呈递给StringTie,StringTie可以利用merge的数据重新估算转录本的丰度,还能额外的提供转录本reads数量的数据给下一步的ballgown
最后一步:Ballgown从上一步获得所有转录本及其丰度,根据实验条件进行分类统计
3. 实战
3.1 将reads比对到参考基因组
for i in {188044,188104,188234,188245,188257,188273,188337,188383,188401,188428,188454,204916};do
hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran -1 chrX_data/samples/ERR${i}_chrX_1.fastq.gz -2 chrX_data/samples/ERR${i}_chrX_2.fastq.gz -S ERR${i}_chrX.sam
done
- -p 开8线程
- --dta Reports alignments tailored for transcript
- -x 后面接文件
- -1 -2 文件一文件二
-
-S 输出为sam 文件
 image.png
image.png - 总共比对了1321477条,9.21%一次都没有比对,78.26%比对到一次,12.52%比对大于一次
- 没有匹配上的9.21%,不按照顺序再匹配,有3.45%匹配上了
- 之后把剩余部分用单端数据进行比对,51.02%没有比对上,40.36%比对了一次,8.62%大于一次
-
总共比对到95.46%
 image.png
image.png
3.2 将SAM文件转换为BAM
for i in {188044,188104,188234,188245,188257,188273,188337,188383,188401,188428,188454,204916};do
samtools sort -@ 8 -o ERR${i}_chrX.bam ERR${i}_chrX.sam
done
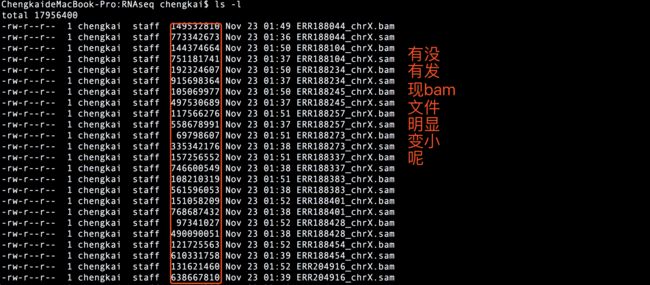
3.3 组装转录本并定量表达基因
for i in {188044,188104,188234,188245,188257,188273,188337,188383,188401,188428,188454,204916};do
stringtie -p 8 -G chrX_data/genes/chrX.gtf -o ERR${i}_chrX.gtf -l ERR${i} ERR${i}_chrX.bam
done

3.4 把所有转录本合并
stringtie --merge -p 8 -G chrX_data/genes/chrX.gtf -o stringtie_merged.gtf chrX_data/mergelist.txt
3.5 重新组装转录本并估算基因表达丰度,并为ballgown创建读入文件
for i in {188044,188104,188234,188245,188257,188273,188337,188383,188401,188428,188454,204916};do
stringtie -e -B -p 8 -G stringtie_merged.gtf -o ballgown/ERR${i}/ERR${i}_chrX.gtf ERR${i}_chrX.bam
done

3.6 R的安装与导入
source("https://bioconductor.org/biocLite.R")
biocLite("ballgown")
source("https://bioconductor.org/biocLite.R")
biocLite("genefilter")
source("https://bioconductor.org/biocLite.R")
biocLite("devtools")
install.packages(dplyr)
- 装RSkittleBrewer有点小问题,如果没报错,那恭喜,如果报错用下面一条
devtools::install_github('alyssafrazee/RSkittleBrewer', force=TRUE)
3.7 R语言脚本
library(RSkittleBrewer)
library(ballgown)
library(genefilter)
library(dplyr)
library(devtools)
# 加载样品的表型数据
pheno_data<-read.csv("/Users/chengkai/Desktop/file/learn/RNAseq/chrX_data/geuvadis_phenodata.csv")
# dataDir指的是数据储存的地方,samplePattern则依据样本的名字来,pheno_data则指明了样本数据的关系,这个里面第一列样本名需要和ballgown下面的文件夹的样本名一样,不然会报错错
bg_chrX=ballgown(dataDir = "/Users/chengkai/Desktop/file/learn/RNAseq/ballgown",samplePattern = "ERR",pData = pheno_data)
# 这里滤掉了样本间差异少于一个转录本的数据
bg_chrX_filt = subset(bg_chrX,"rowVars(texpr(bg_chrX)) >1",genomesubset=TRUE)
# 比较男和女的基因表达差异
results_transcripts = stattest(bg_chrX_filt, feature="transcript",covariate="sex",adjustvars = c("population"), getFC=TRUE, meas="FPKM")
# 确认组间有差异的基因
results_genes = stattest(bg_chrX_filt, feature="gene", covariate="sex", adjustvars = c("population"), getFC=TRUE, meas="FPKM")
# 对结果增加基因名和基因ID
results_transcripts = data.frame(geneNames=ballgown::geneNames(bg_chrX_filt), geneIDs=ballgown::geneIDs(bg_chrX_filt), results_transcripts)
# 按照P值排序(从小到大)
results_transcripts = arrange(results_transcripts,pval)
results_genes = arrange(results_genes,pval)
# 把结果写到csv文件
write.csv(results_transcripts, "chrX_transcript_results.csv", row.names=FALSE)
write.csv(results_genes, "chrX_gene_results.csv", row.names=FALSE)
# 赋予调色板五个指定颜色
tropical= c('darkorange', 'dodgerblue', 'hotpink', 'limegreen', 'yellow')
palette(tropical)
# 提取FPKM值
fpkm = texpr(bg_chrX,meas="FPKM")
#方便作图将其log转换,+1是为了避免出现log2(0)的情况
fpkm = log2(fpkm+1)
# 作图
boxplot(fpkm,col=as.numeric(pheno_data$sex),las=2,ylab='log2(FPKM+1)')
# 就单个转录本的查看在样品中的分布
ballgown::transcriptNames(bg_chrX)[12]
ballgown::geneNames(bg_chrX)[12]
plot(fpkm[12,] ~ pheno_data$sex, border=c(1,2), main=paste(ballgown::geneNames(bg_chrX)[12],' : ', ballgown::transcriptNames(bg_chrX)[12]),pch=19, xlab="Sex", ylab='log2(FPKM+1)')
points(fpkm[12,] ~ jitter(as.numeric(pheno_data$sex)), col=as.numeric(pheno_data$sex))
#查看某一基因位置上所有的转录本
plotTranscripts(ballgown::geneIDs(bg_chrX)[1729], bg_chrX, main=c('Gene XIST in sample ERR188234'), sample=c('ERR188234'))
# 以性别为区分查看基因表达情况
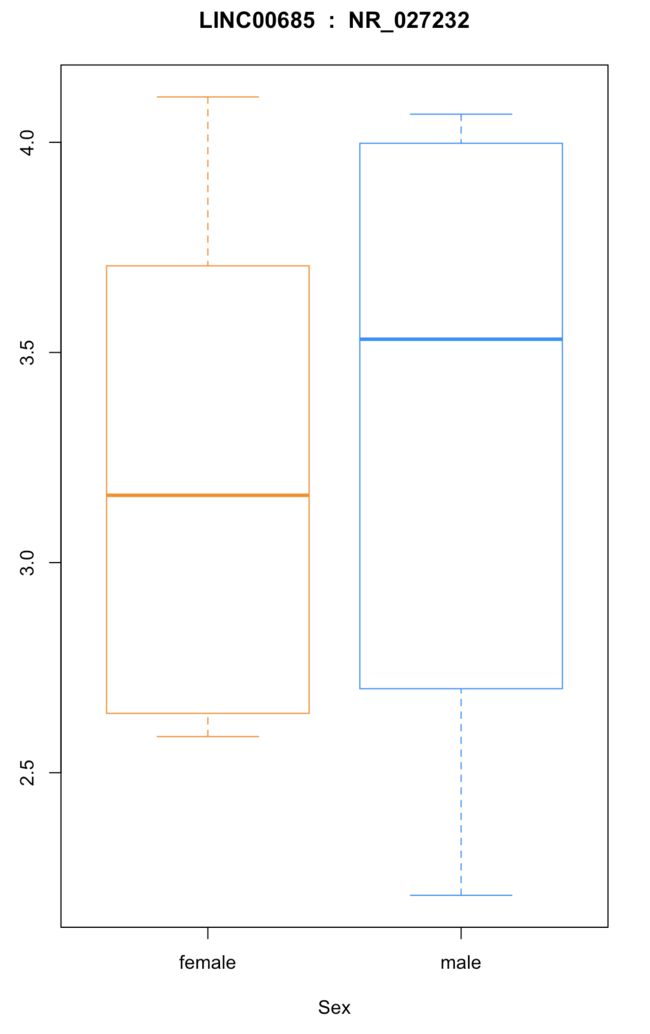
plotMeans('MSTRG.575', bg_chrX_filt,groupvar="sex",legend=FALSE)
3.8 数据分析
-
pheno_data:表型数据
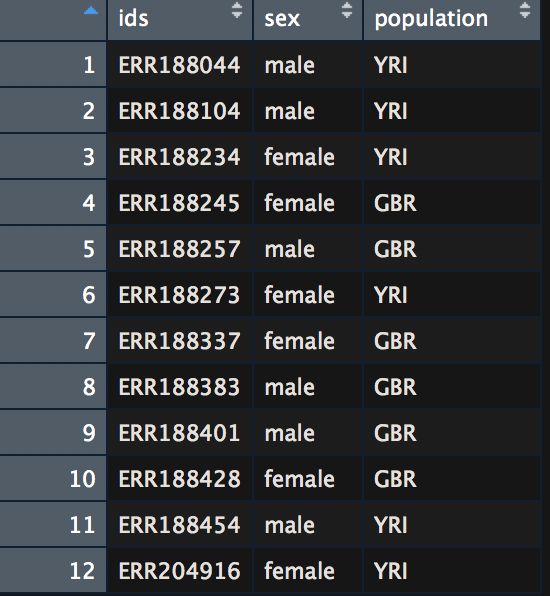 image.png
image.png -
results_transcripts:比较男和女的基因表达差异
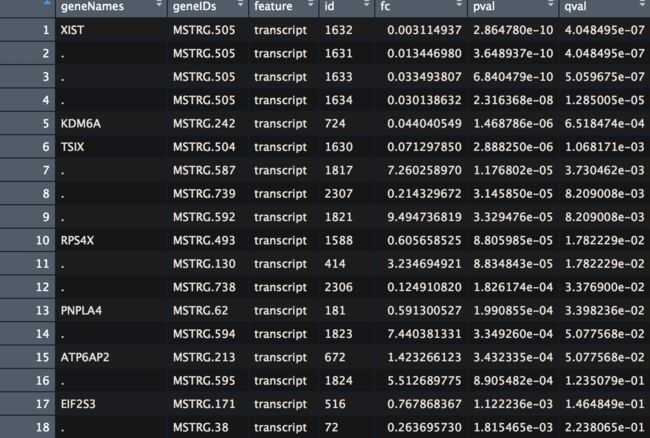 image.png
image.png -
results_genes:确认组间有差异的基因
 image.png
image.png -
fpkm 作图(男的为蓝色,女的为橙色)
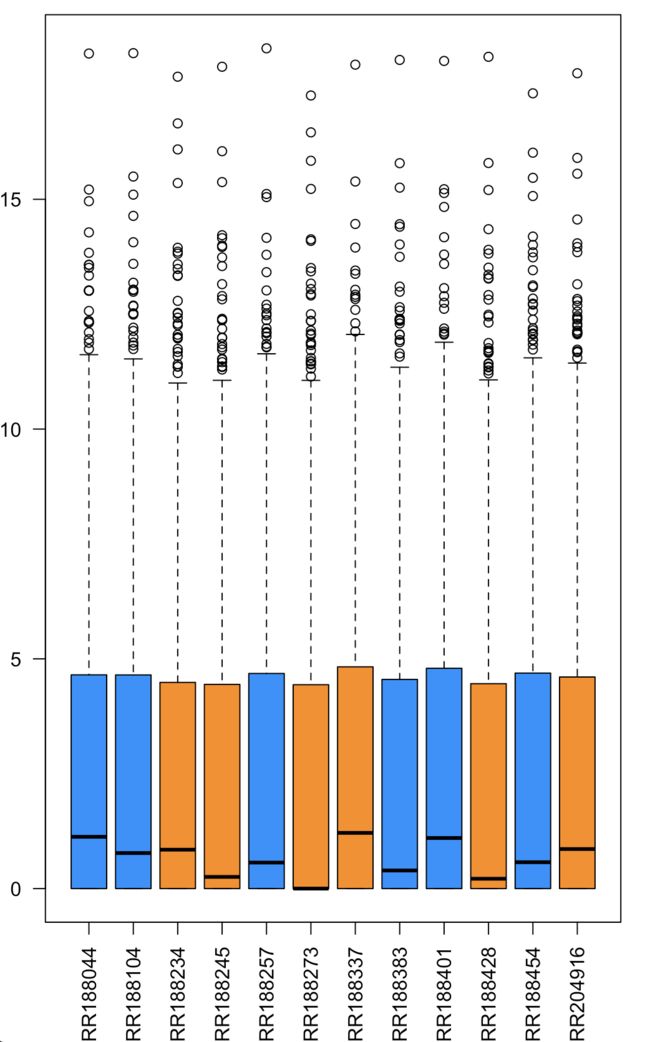 image.png
image.png -
就单个转录本的查看在样品中的分布
 image.png
image.png
-
查看某一基因位置上所有的转录本
 image.png
image.png -
以性别为区分查看基因表达情况
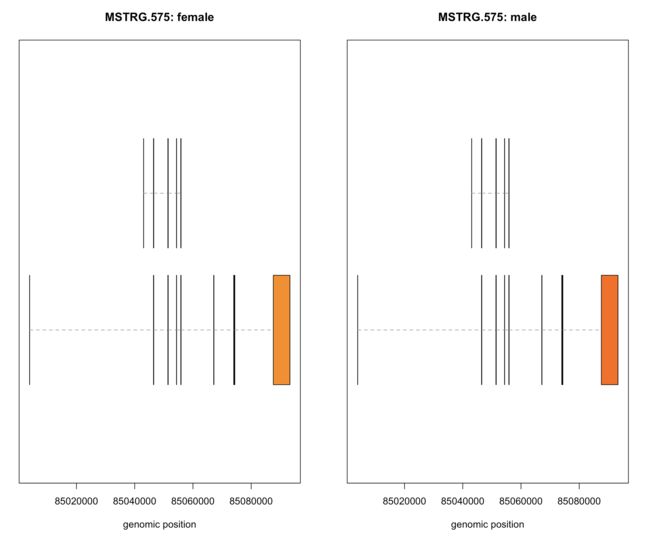 image.png
image.png
3.9 结论
-
软件的比对效果非常好,95%比对上啦
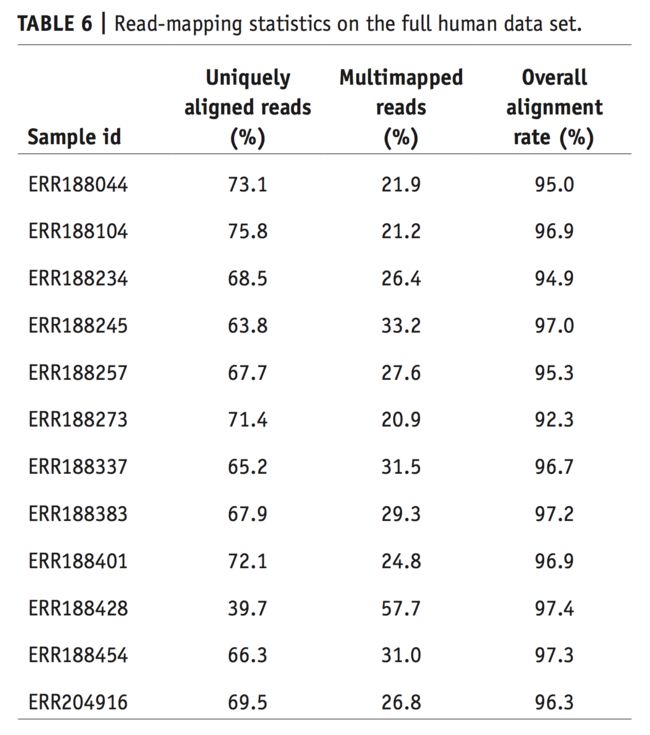 image.png
image.png -
转录组组装情况一览
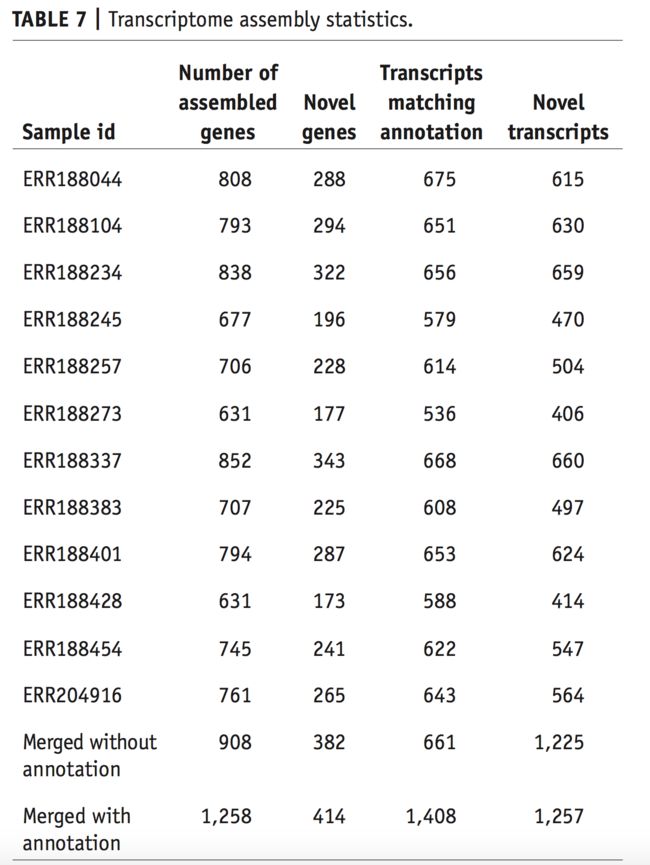 image.png
image.png -
p值分布
 image.png
image.png
参考文献
- http://www.jianshu.com/p/1f5d13cc47f8(徐爷大神)
- https://www.nature.com/articles/nprot.2016.095.pdf