@我的博客:有味
导读
目前用于代谢组学数据的液相色谱和质谱分析的工具只涵盖有限的处理步骤,而在线工具很难以可编程的方式使用。这篇文章作者给我们分享了一个可以用于代谢物自动鉴定的工具包,可以让我们轻松驾驭从质谱数据的获到末端数据的统计结果着一整个流程。
MAIT(Metabolite Automatic Identification Toolkit )致力于改进峰值注释阶段,并提供必要的工具来验证统计分析结果。整个流程下来可以得到统计分析的结果,峰注释的结果以及代谢物鉴定的结果。
文章介绍
Title: An R package to analyse LC/MS metabolomic data: MAIT (Metabolite Automatic Identification Toolkit)[1]
译名:分析液相色谱质谱代谢组学数据R包-MAIT
期刊:Bioinformatics
IF:5.481
作者:Francesc Fernández-Albert
单位:Bioinformatics and Biomedical Signals Laboratory(简称B2B实验室),位于西班牙巴塞罗那的加泰罗尼亚理工大学
实验室网站:B2B
主要研究领域:「Biomedical Engineering, Bioinformatics, Machine Learning and Statistics」
Pipline
LC/MS代谢组数据的主要处理步骤包括:峰值检测、峰值注释和统计分析三大步骤。
- 峰值检测:峰值检测步骤,毫无疑问就是检测LC/MS样本文件中的色谱峰和质谱峰;
- 峰值注释:通过增加数据集中的化学和生物信息背景,更好地鉴定代谢组学样品中的代谢物;

- 统计分析:这一步是通过代谢组学数据获得具有重要生物学意义的代谢物特征值的基础。
软件安装
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("MAIT", version = "3.8")
示例数据及加载数据
用的是参考文献的数据,内置在XCMS包里面。该数据包含两组数据,一组是小鼠脂肪酸酰胺水解酶基因敲除(KO组),一组是野生型小鼠(WT组),每组包括6个脊髓液样品,那么对于MAIT包来说,数据的存放需要符合一定的规范,不同分组需设置不同的文件夹,然后存放于一个总的分析文件夹下,如下图所示:
峰值检测
一旦数据加载好,就可以进行峰值检测啦
library(MAIT)
library(faahKO)
cdfFiles<-system.file("cdf", package="faahKO", mustWork=TRUE)
MAIT <- sampleProcessing(dataDir = cdfFiles, project = "MAIT_Demo",
snThres=2,rtStep=0.03)
# 由于这里用的是示例数据,如果是自己的数据那么dataDir后接的就是自己设置的文件夹,project是自己要存放的结果文件的目录
# snThres表示的是信噪比参数的设置,rtStep生成峰文件的步长
MAIT
# A MAIT object built of 12 samples
# The object contains 6 samples of class KO
# The object contains 6 samples of class WT
- 执行sampleProcessing函数后,软件开始检测峰值,根据样本分组,校正保留时间等**
- 结果文件就是存放在MAIT里,它包括检测到的峰值,分组信息,以及每组包括多少文件。具体信息可以通过命令
summary(MAIT)来获得
峰注释
接下来是峰注释的第一步,通过函数peakAnnotation()来实现,如果adductTable参数没有事先设定,那么默认的就是以正离子加和离子形式进行,除非该参数值设定为”negAdducts”。
MAIT <- peakAnnotation(MAIT.object = MAIT,corrWithSamp = 0.7,
corrBetSamp = 0.75,perfwhm = 0.6)
# corrWithSamp参数是样品内峰值的相关系数,corrBetSamp则是样品间的一个相关系数
# perfwhm参数是根据保留时间来区分两个峰值
rawData(MAIT) # 结果包括相关的峰、质谱图以及注释结果
# $xsaFA
# An "xsAnnotate" object!
# With 735 groups (pseudospectra)
# With 12 samples and 1261 peaks
# Polarity mode is set to: positive
# Using automatic sample selection
# Annotated isotopes: 104
# Annotated adducts & fragments: 72
# Memory usage: 4.35 MB
注:这里有必要写点内容解释下perfwhm参数到底是什么意思
在液相色谱质谱峰检测过程中,如果两个峰是共流出的,那么我们有一个定义,那就是其要落在一个范围内,什么范围呢,就是
Rt_med +/- FWHM * perfwhm,Rt_med是保留时间的中位数,FWHM(Full Width at Half Maximum,翻译为半峰全宽[2]),我们暂且假设一个质谱峰为高斯峰,那么当等于最高峰强度值的一半时,X轴两点之间的距离就叫FWHM,那么perfwhm参数的设定就是FWHM的百分比。

统计分析
在进行完第一个峰值注释阶段之后,我们想知道不同分组之间的哪些代谢物特征值是有差异的。因此,我们运行函数spectralSigFeatures()
MAIT<- spectralSigFeatures(MAIT.object = MAIT,pvalue=0.05,
p.adj="none",scale=FALSE)
# pvalue设置组间比较的阈值
# p.adjust选择p值校正方法
# scale是一个逻辑值,分析前是否要进行标度化
summary(MAIT) # 查看结果
# A MAIT object built of 12 samples and 1261 peaks. No peak aggregation technique has been applied
# 56 of these peaks are statistically significant
# The object contains 6 samples of class KO
# The object contains 6 samples of class WT
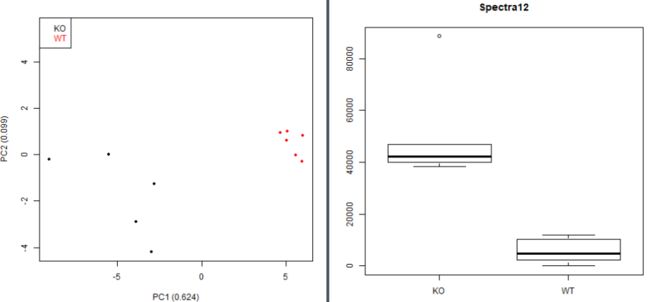
统计作图
plotBoxplot(MAIT)
plotHeatmap(MAIT)
MAIT<-plotPCA(MAIT,plot3d=FALSE)
MAIT<-plotPLS(MAIT,plot3d=FALSE)
PLSmodel <- model(MAIT, type = "PLS")
PCAmodel <- model(MAIT, type = "PCA") # 这里就是对统计分析得到的56个在组间差异显著的峰值进行可视化,有箱线图,热图,PCA和PLS图
PLSmodel # PLS模型分析还可以根据自举法抽样将代谢物用于组间的预测。可以看到预测结果比较准确
resultsPath(MAIT) # 另外还可以根据resultPath获得结果所存放的目录在哪里
生物转化
在物质鉴定之前,能生物转换可以提升peak的注释,有助于结果的呈现和解释,MAIT包使用一个默认的生物转换表,但是用户可以自己自行定义,并通过使用生物表函数加载变量
Biotransformations(MAIT.object = MAIT, peakPrecision = 0.005)
# peakPrecision参数用于设置实际的峰质量值和生物转换表差异的允许范围
举个栗子
从MAIT默认表构建用户定义的生物转换表或添加新的生物转换非常简单,比如我们想自己添加一个新的加和物到默认的生物转换表里
data(MAITtables)
myBiotransformation<-c("custom_biotrans",105.0)
myBiotable<-biotransformationsTable
myBiotable[,1]<-as.character(myBiotable[,1])
myBiotable<-rbind(myBiotable,myBiotransformation)
myBiotable[,1]<-as.factor(myBiotable[,1])
tail(myBiotable) # 查看添加了新的加和物的生物转换表,只看最后几行
# NAME MASSDIFF
# 45 glucuronide conjugation 176.0321
# 46 hydroxylation + glucuronide 192.027
# 47 GSH conjugation 305.0682
# 48 2x glucuronide conjugation 352.0642
# 49 [C13] 1.0034
# 50 custom_biotrans 105
可以看到,其实就是在原来的生物转换表的基础上新加了一行,很简单对不对
物质鉴定
一旦完成了生物转化注释步骤,重要的特征值通过更加准确的注释得到富集。biotransformation()函数执行的注释过程永远不会取代其他函数已经完成的峰值注释。MAIT认为peak注释是互补的;因此,当检测到新的注释时,可以将其添加到当前的peak注释结果中,并启动鉴定功能,识别数据中具有统计学意义的特征值所对应的代谢物
MAIT <- identifyMetabolites(MAIT.object = MAIT, peakTolerance = 0.005)
metTable<-metaboliteTable(MAIT)
head(metTable)
默认情况下,identifyMetabolites()函数在默认代谢物数据库中寻找重要的特征值的峰值。参数peakTolerance定义峰值和数据库中化合物匹配的阈值。默认情况下设定为0.005的质荷比。为了使结果易于检查,identifyMetabolites()函数创建了一个包含重要的特征值和可能的代谢物鉴定结果表
验证
最后,我们将使用函数Validation()来检查重要特征值的预测值。与Validation()函数输出相关的所有信息都保存在名为“Validation”的文件夹中的目录中。创建了两个可以显示总体和每组的分类比率的箱线图,以及迭代对应的混淆矩阵结果
MAIT <- Validation(Iterations = 20, trainSamples= 3, MAIT.object = MAIT)
# Iterations设定迭代次数,trainSamples设置选择多样样品来做验证
summary(MAIT) # 查看结果
# A MAIT object built of 12 samples and 1261 peaks. No peak aggregation technique has been applied
# 56 of these peaks are statistically significant
# The object contains 6 samples of class KO
# The object contains 6 samples of class WT
# The Classification using 3 training samples and 20 Iterations gave the results:
# KNN PLSDA SVM
# mean 1 1 1
# standard error 0 0 0
可以看到分类的比例为100%,说明这些重要的代谢物特征值可以很好的区分两个分组的样品
参考
[1] 原文链接
[2] FWHM半峰全宽