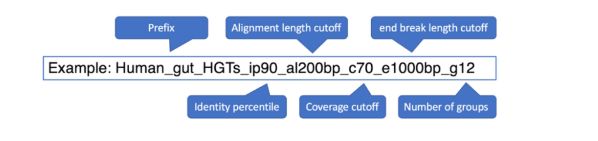
- 【LeetCode】215.数组中的第K个最大元素(三种方法,九个思路的代码实现,java格式)
Hi丶ImViper
LeetCode算法与数据结构算法数据结构java快速排序
题目题目链接解析这道题据说是面试的高频考题,同时也是基础算法的应用。方法一:暴力解法题目要求我们找到“数组排序后的第k个最大的元素,而不是第k个不同的元素”,语义是从右边往左边数第k个元素(从11开始),那么从左向右数是第几个呢,我们列出几个找找规律就好了。一共6个元素,找第2大,索引是4;一共6个元素,找第4大,索引是2。因此,升序排序以后,目标元素的索引是len-k。这是最简单的思路,如果只答
- LeetCode:215 数组中第k个最大元素 优先队列 / 堆
AkagiSenpai
LeetCode数据结构leetcode数据结构优先队列堆
在未排序的数组中找到第k个最大的元素。请注意,你需要找的是数组排序后的第k个最大的元素,而不是第k个不同的元素。示例1:输入:[3,2,1,5,6,4]和k=2输出:5示例2:输入:[3,2,3,1,2,4,5,5,6]和k=4输出:4说明:你可以假设k总是有效的,且1≤k≤数组的长度。来源:力扣(LeetCode)链接:https://leetcode-cn.com/problems/kth-l
- 力扣215.数组中第k个最大元素
失落的换海迷风
c++
在未排序的数组中找到第k个最大的元素。请注意,你需要找的是数组排序后的第k个最大的元素,而不是第k个不同的元素。classSolution{public:intfindKthLargest(vector&nums,intk){//第一种方法:优先级队列////建立大堆//priority_queuep(nums.begin(),nums.end());////出前k-1个元素//for(inti=
- 力扣215. 数组中的第K个最大元素
hyssop2019
算法leetcode算法排序算法
题目描述给定整数数组nums和整数k,请返回数组中第k个最大的元素。请注意,你需要找的是数组排序后的第k个最大的元素,而不是第k个不同的元素。你必须设计并实现时间复杂度为O(n)的算法解决此问题。示例1:输入:[3,2,1,5,6,4],k=2输出:5示例2:输入:[3,2,3,1,2,4,5,5,6],k=4输出:4提示:1cursor){returnpartition(nums,cursor+
- 手撕力扣之排序:排序数组、数组中的逆序对、排序链表、最小的k个数、数组中的第K个最大元素、前 K 个高频元素、根据身高重建队列、最大数、下一个排列、下一个更大元素 III、最大交换、字典序的第K小数字
weixin_39770712
数据结构与算法数据结构排序算法算法
力扣912.排序数组给你一个整数数组nums,请你将该数组升序排列。方法一:归并排序classSolution{public:vectortmp;voidmergeSort(vector&nums,intl,intr){if(l>=r)return;intmid=(l+r)>>1;mergeSort(nums,l,mid);mergeSort(nums,mid+1,r);inti=l,j=mid+
- leetcode:206.反转链表
uncle_ll
编程练习-Leetcodeleetcode链表反转递归迭代算法训练营
206.反转链表来源:力扣(LeetCode)链接:https://leetcode.cn/problems/reverse-linked-list给你单链表的头节点head,请你反转链表,并返回反转后的链表。示例1:输入:head=[1,2,3,4,5]输出:[5,4,3,2,1]示例2:输入:head=[1,2]输出:[2,1]示例3:输入:head=[]
- 单端口和双单口RAM的实现
wangn1633
Verilogverilog
单端口和双单口RAM的verilog实现概念:1单端口:读写数据共用一个地址线,一个时钟沿只能进行读或者写;2伪双端口:写数据和读数据有自己的地址、时钟、读写使能信号;也就是一组端口只能写,一组端口只能读。(读写数据也可共用一个clk,为同步伪双端口ram)3真双端口:一组端口可读可写,另一组端口也可读可写。(若这两组端口共用一个clk,为同步真双端口ram。若每组有每组的clk,为异步真双端口r
- 力扣p234:回文链表
&可 乐
力扣+牛客练习题链表javaleetcode
题目:回文链表题目描述:请判断一个链表是否为回文链表思路1:根据回文结构的性质,直接反转整个链表,然后比较两个链表是否相同这种方法不推荐用,因为要反转链表要开辟新空间,空间复杂度会大于O(1),不建立新链表的话会浅拷贝,出错。思路2:找到链表中间结点。反转后半部分链表,这样不用开辟新空间,满足时间复杂度和空间复杂度的要求。代码://回文链表publicstaticbooleanisPalindro
- jolt transform (json to json) 嵌套数组进行json格式转换
安静的数据流
jolt
输入的json格式:[{"status":"success","result":[{"cashierUid":1111,"items":[{"name":"name1"}]},{"cashierUid":2222,"items":[{"name":"name2"},{"name":"name22"}]}],"pageSize":2},{"status":"success","result":[{"
- leetcode 215. 数组中的第 K个最 大的元素(堆排序,C语言)
Oh?Geostatistics…
算法与数据结构数据结构排序算法堆排序
数组排序后的第k个最大的元素,而不是第k个不同的元素快速排序intcmp(constvoid*a,constvoid*b){return*(int*)b-*(int*)a;}intfindKthLargest(int*nums,intnumsSize,intk){qsort(nums,numsSize,sizeof(int),cmp);returnnums[k-1];}堆排序```c/*交换*/v
- 力扣215.数组中的第K个最大元素
Lucky小黄人
数据结构算法leetcode快速排序排序算法
215.数组中的第K个最大元素在未排序的数组中找到第k个最大的元素。请注意,你需要找的是数组排序后的第k个最大的元素,而不是第k个不同的元素。示例1:输入:[3,2,1,5,6,4]和k=2输出:5示例2:输入:[3,2,3,1,2,4,5,5,6]和k=4输出:4说明:你可以假设k总是有效的,且1≤k≤数组的长度。思路一:排序后取值复杂度分析:时间复杂度:排序时间为O(nlogn),所以时间为O
- LeetCode215.数组中的第K个最大元素 java使用小顶堆求解
patientany
java开发语言
JAVA实现小顶堆手撕小顶堆定义堆中的成员变量提供构造方法建堆下潜交换堆的尾部添加元素上浮获取堆顶元素替换堆顶元素删除指定元素删除堆顶元素回到题目具体步骤上代码手撕小顶堆在java中实现小顶堆定义堆中的成员变量这里首先先定义堆中的数据,在这里我使用了整数数组表示整个堆。size表示堆的大小,默认也就是数组的长度。int[]array;intsize;提供构造方法对于堆的初始化,由传进来的数组实现对
- 网络实验操作-VLAN
会员果汁
网络
实验目的了解VLAN的作用和配置。基础实验需求:所有PC在相同网段,但只有PC1和PC3可以互通,PC2和PC4可以互通配置思路将可以互通的PC放到相同的VLAN中即可。配置过程某一个switch的配置:#interfaceGigabitEthernet0/0/1portlink-typetrunkporttrunkallow-passvlan1020#interfaceGigabitEthern
- jolt json to json mapping第一篇
chizawu5345
jsonjava
demo解读本文的主要目的是整理梳理对于joltjson的使用,主要使用场景就是jsonmapping模式的含义shift清空后输出default直接输出,类似于增量*匹配所有&取key值出现在value里例子:&=&0当前层级&1向上1级value里没有能力获取key值但是可以通过*,$配合使用$取key值出现在key里同上@取value值出现在key里例子:@(3,clientId)@取val
- 配置OSPF与BFD联动
IT_社恐刘某
服务器运维
目录BFD简介配置BFD目的BFD优点受益OSPF简介定义OSPF优点实验组网需求配置思路配置步骤1.配置各接口所属的VLAN2.配置各VLANIF接口的IP地址3.配置OSPF基本功能4.配置OSPF与BFD联动5.检查配置结果BFD简介双向转发检测BFD(BidirectionalForwardingDetection)是一种全网统一的检测机制,用于快速检测、监控网络中链路或者IP路由的转发连
- 莫名锁表? --- mysql的事务隔离级别
程序员小软
mysql数据库java
前言系统响应超时系统访问数据库特别慢莫名提示锁等待超时数据库锁表事务长时间等锁,直到超时以上问题都可能是事务锁表导致的问题今天测试反馈系统批量处理莫名提示锁等待超时,再次操作查看数据库事务确实存在等锁情况,甚至死锁。刚开始是偶尔出现,后来一直就是死锁,导致其他操作也操作不了。刚开始发现数据库中操作插入的时候会进入锁等待怀疑是这张表中主键自增导致的锁表,于是将表改为指定主键,问题依然存在。后来想起来
- linux操作速查
程序员小软
linux运维linux服务器运维
功能创建新用户并赋予root权限切记在root身份下操作查看用户列表cat/etc/passwd创建新用户useradd为账户设置密码passwd赋予root权限编辑/etc/sudoers文件添加一行ALL=(ALL)ALL验证sudo-l#查看当前用户可使用的命令,仅限root用户可用sudo-i#命令切换到root权限问题处理处理端口被占用:netstat-ano|find“80”//列出端
- ⚡️Jolt -- 通过JSON配置来处理复杂数据转换的工具
程序员小软
工具javajson数据处理
简介:一个能够通过JSON配置(特定的语法)来处理复杂数据转换的工具。比如将API响应转换为内部系统所需的格式,或者处理来自不同来源的数据结构差异。例如,将嵌套的JSON结构扁平化,或者重命名字段,合并多个字段等。名称含义:JSON+Bolt:JSON是工具处理的数据格式。Bolt(闪电)象征快速和高效,暗示Jolt能够像闪电一样快速完成JSON数据的转换。Bolt的logo就是一个闪电⚡️标志在
- 006 python-if条件
梅洪
pythonpython服务器开发语言
Pythonif条件教学设计一、教学目标了解if语句的基本结构和执行逻辑。掌握if-else和if-elif-else语句的使用方法。能够运用条件判断解决实际问题,如分数判断、用户登录等。理解if语句中的比较运算符和逻辑运算符的作用。二、教学重点if语句的基本语法if-else语句if-elif-else语句逻辑运算符and、or、not在if语句中的应用三、教学难点多重if-elif-else结
- 答题卡图像识别 需求分析、市场分析和技术实现
weixin_34037977
人工智能开发工具json
答题卡图像识别需求分析、市场分析和技术实现一、需求分析一、以接口的方式开发此需求:1:接收图片以上传的方式把图片发送到接口。2:识别图片接口接收到图片后,进行图像识别。3:返回数据返回识别后的JSON格式数据。二、答题卡图片识别的具体要求:图片是通过手机、相机、扫描仪等设备拍照而来,其中手机、相机拍出的照片会出现像素低、图像不正、聚焦不清楚等问题;1:图片只要是人眼能看清楚的即可完成识别;2:80
- 神经网络模型压缩&实例教程—非结构化剪枝
程序先锋
《python深度学习》笔记神经网络剪枝深度学习
目录1.导包&定义一个简单的网络2.获取网络需要剪枝的模块3.模块剪枝(核心)3.1随机剪枝weight3.2L1范数剪枝bias4.总结最先进的深度学习技术依赖于难以部署的过度参数化模型。相反,已知生物神经网络使用高效的稀疏连接。为了在不牺牲准确性的情况下减少内存、电池和硬件消耗,通过减少模型中的参数数量来确定压缩模型的最佳技术是很重要的。这反过来又允许您在设备上部署轻量级模型,并通过设备上的私
- RPC远程调用框架Dubbo
Czi橙
rpcdubbo网络协议javanacosspringcloud微服务
一、分布式服务调用_什么是RPCRPC(RemoteProcedureCall)远程过程调用,它是一种通过网络从远程计算机程序上请求服务。大白话理解就是:RPC让你用别人家的东西就像自己家的一样。RPC两个作用:屏蔽远程调用跟本地调用的区别,让我们感觉就是调用项目内的方法隐藏底层网络通信的复杂性,让我们更加专注业务逻辑。常用的RPC框架RPC是一种技术思想而非一种规范或协议。常见RPC技术和框架:
- elementui 组件基本颜色的修改
怡宝丶加冰
vue+elementuielementui前端javascript
再用elementui中的组件时根据项目的不同主题色也不一样,这里是对一些常用组件基础颜色的修改,可以直接用,根据主题色的不同直接替换--theme_color变量的值就行创建一个css文件/*全局样式*/html,body{--theme_color:#0C871B;}/*primary按钮样式*/.el-button--primary{background-color:var(--theme_
- dhtmlxGantt 甘特图 一行展示多条数据
怡宝丶加冰
甘特图
效果如图:后台拿到数据处理之后如图:含义:如上图所示,如果一行需要展示多个需要给父数据的那条添加render:split属性,子数据的parent为父数据的Id即可切记父数据的id别为0为0时会出现错乱因为有些小伙伴提出分段展示的数据结构还是有点问题,下面展示一个完整的demoimport{gantt}from'dhtmlx-gantt';import"dhtmlx-gantt/codebase/
- 【python双目标定轮椅】基于python的双目标定
迟钝皮纳德
pythonopencv计算机视觉
代码部分话不多说直接上代码:新建文件getdata.pyimportcv2importosid_image=0#图片的IDcamera=cv2.VideoCapture(1)#找到棋盘格的标准criteria=(cv2.TERM_CRITERIA_EPS+cv2.TERM_CRITERIA_MAX_ITER,30,0.001)camera.set(cv2.CAP_PROP_FRAME_WIDTH,
- Docker自动安装、启动、接入、结束ros-melodic-desktop-full的脚本
QxAIRobot
robotshell脚本dockerROSlinux机器人ubuntu
脚本#!/bin/bash#Ascripttostartros:melodic-desktop-fullindocker.#Youneedtoinstallthelatestdockerfirst.#Author:liuqixuan.cn#Email:
[email protected]#set-xRES_NAME=rosTAG=melodic-desktop-fullif[[-z`whichdoc
- Python 中的离线语音转文本
无水先生
语音编程人工智能综合python开发语言
Python中的离线语音转文本一、说明 写作、编码、写博客、办公室工作、文档、报告都需要一个人在键盘上打字。这会导致健康问题,如腕管综合症、手和手指疼痛等。我非常了解这种痛苦。这是用于创建自己的离线运行的听写程序的Python代码。只需对着耳机的麦克风说话,它就会将您的话转换为文本并将其保存在文本文件中。二、安装 您将需要安装Python库—vosk、pyaudio。 Vosk是一个语音识别
- 毕设分享 大数据B站数据分析可视化系统
bee_dc
毕业设计毕设大数据
文章目录0前言1项目运行效果2设计原理数据处理方案可视化呈现方案综合得分计算指标综合得分漏斗图游客画像完成度三连排行榜点赞、投币、收藏与白嫖的比例分析3最后0前言这两年开始毕业设计和毕业答辩的要求和难度不断提升,传统的毕设题目缺少创新和亮点,往往达不到毕业答辩的要求,这两年不断有学弟学妹告诉学长自己做的项目系统达不到老师的要求。为了大家能够顺利以及最少的精力通过毕设,学长分享优质毕业设计项目,今天
- 毕业设计项目 大数据B站数据分析可视化系统
bee_dc
毕业设计毕设大数据
文章目录0前言1项目运行效果2设计原理数据处理方案可视化呈现方案综合得分计算指标综合得分漏斗图游客画像完成度三连排行榜点赞、投币、收藏与白嫖的比例分析3最后0前言这两年开始毕业设计和毕业答辩的要求和难度不断提升,传统的毕设题目缺少创新和亮点,往往达不到毕业答辩的要求,这两年不断有学弟学妹告诉学长自己做的项目系统达不到老师的要求。为了大家能够顺利以及最少的精力通过毕设,学长分享优质毕业设计项目,今天
- Dockerfile及Docker-compose Yaml
Darklord.W
dockerdockerdockerfiledocker-composeyaml
Dockerfile一、结构:基础镜像信息维护者信息镜像操作指令容器启动时执行指令FROM指明构建的新镜像是来自于哪个基础镜像,例如:FROMcentos:6MAINTAINER指明镜像维护着及其联系方式(一般是邮箱地址),例如:MAINTAINEREdisonZhou不过,MAINTAINER并不推荐使用,更推荐使用LABEL来指定镜像作者,例如:LABELmaintainer="xxxxx.c
- mysql主从数据同步
林鹤霄
mysql主从数据同步
配置mysql5.5主从服务器(转)
教程开始:一、安装MySQL
说明:在两台MySQL服务器192.168.21.169和192.168.21.168上分别进行如下操作,安装MySQL 5.5.22
二、配置MySQL主服务器(192.168.21.169)mysql -uroot -p &nb
- oracle学习笔记
caoyong
oracle
1、ORACLE的安装
a>、ORACLE的版本
8i,9i : i是internet
10g,11g : grid (网格)
12c : cloud (云计算)
b>、10g不支持win7
&
- 数据库,SQL零基础入门
天子之骄
sql数据库入门基本术语
数据库,SQL零基础入门
做网站肯定离不开数据库,本人之前没怎么具体接触SQL,这几天起早贪黑得各种入门,恶补脑洞。一些具体的知识点,可以让小白不再迷茫的术语,拿来与大家分享。
数据库,永久数据的一个或多个大型结构化集合,通常与更新和查询数据的软件相关
- pom.xml
一炮送你回车库
pom.xml
1、一级元素dependencies是可以被子项目继承的
2、一级元素dependencyManagement是定义该项目群里jar包版本号的,通常和一级元素properties一起使用,既然有继承,也肯定有一级元素modules来定义子元素
3、父项目里的一级元素<modules>
<module>lcas-admin-war</module>
<
- sql查地区省市县
3213213333332132
sqlmysql
-- db_yhm_city
SELECT * FROM db_yhm_city WHERE class_parent_id = 1 -- 海南 class_id = 9 港、奥、台 class_id = 33、34、35
SELECT * FROM db_yhm_city WHERE class_parent_id =169
SELECT d1.cla
- 关于监听器那些让人头疼的事
宝剑锋梅花香
画图板监听器鼠标监听器
本人初学JAVA,对于界面开发我只能说有点蛋疼,用JAVA来做界面的话确实需要一定的耐心(不使用插件,就算使用插件的话也没好多少)既然Java提供了界面开发,老师又要求做,只能硬着头皮上啦。但是监听器还真是个难懂的地方,我是上了几次课才略微搞懂了些。
- JAVA的遍历MAP
darkranger
map
Java Map遍历方式的选择
1. 阐述
对于Java中Map的遍历方式,很多文章都推荐使用entrySet,认为其比keySet的效率高很多。理由是:entrySet方法一次拿到所有key和value的集合;而keySet拿到的只是key的集合,针对每个key,都要去Map中额外查找一次value,从而降低了总体效率。那么实际情况如何呢?
为了解遍历性能的真实差距,包括在遍历ke
- POJ 2312 Battle City 优先多列+bfs
aijuans
搜索
来源:http://poj.org/problem?id=2312
题意:题目背景就是小时候玩的坦克大战,求从起点到终点最少需要多少步。已知S和R是不能走得,E是空的,可以走,B是砖,只有打掉后才可以通过。
思路:很容易看出来这是一道广搜的题目,但是因为走E和走B所需要的时间不一样,因此不能用普通的队列存点。因为对于走B来说,要先打掉砖才能通过,所以我们可以理解为走B需要两步,而走E是指需要1
- Hibernate与Jpa的关系,终于弄懂
avords
javaHibernate数据库jpa
我知道Jpa是一种规范,而Hibernate是它的一种实现。除了Hibernate,还有EclipseLink(曾经的toplink),OpenJPA等可供选择,所以使用Jpa的一个好处是,可以更换实现而不必改动太多代码。
在play中定义Model时,使用的是jpa的annotations,比如javax.persistence.Entity, Table, Column, OneToMany
- 酸爽的console.log
bee1314
console
在前端的开发中,console.log那是开发必备啊,简直直观。通过写小函数,组合大功能。更容易测试。但是在打版本时,就要删除console.log,打完版本进入开发状态又要添加,真不够爽。重复劳动太多。所以可以做些简单地封装,方便开发和上线。
/**
* log.js hufeng
* The safe wrapper for `console.xxx` functions
*
- 哈佛教授:穷人和过于忙碌的人有一个共同思维特质
bijian1013
时间管理励志人生穷人过于忙碌
一个跨学科团队今年完成了一项对资源稀缺状况下人的思维方式的研究,结论是:穷人和过于忙碌的人有一个共同思维特质,即注意力被稀缺资源过分占据,引起认知和判断力的全面下降。这项研究是心理学、行为经济学和政策研究学者协作的典范。
这个研究源于穆来纳森对自己拖延症的憎恨。他7岁从印度移民美国,很快就如鱼得水,哈佛毕业
- other operate
征客丶
OSosx
一、Mac Finder 设置排序方式,预览栏 在显示-》查看显示选项中
二、有时预览显示时,卡死在那,有可能是一些临时文件夹被删除了,如:/private/tmp[有待验证]
--------------------------------------------------------------------
若有其他凝问或文中有错误,请及时向我指出,
我好及时改正,同时也让我们一
- 【Scala五】分析Spark源代码总结的Scala语法三
bit1129
scala
1. If语句作为表达式
val properties = if (jobIdToActiveJob.contains(jobId)) {
jobIdToActiveJob(stage.jobId).properties
} else {
// this stage will be assigned to "default" po
- ZooKeeper 入门
BlueSkator
中间件zk
ZooKeeper是一个高可用的分布式数据管理与系统协调框架。基于对Paxos算法的实现,使该框架保证了分布式环境中数据的强一致性,也正是基于这样的特性,使得ZooKeeper解决很多分布式问题。网上对ZK的应用场景也有不少介绍,本文将结合作者身边的项目例子,系统地对ZK的应用场景进行一个分门归类的介绍。
值得注意的是,ZK并非天生就是为这些应用场景设计的,都是后来众多开发者根据其框架的特性,利
- MySQL取得当前时间的函数是什么 格式化日期的函数是什么
BreakingBad
mysqlDate
取得当前时间用 now() 就行。
在数据库中格式化时间 用DATE_FORMA T(date, format) .
根据格式串format 格式化日期或日期和时间值date,返回结果串。
可用DATE_FORMAT( ) 来格式化DATE 或DATETIME 值,以便得到所希望的格式。根据format字符串格式化date值:
%S, %s 两位数字形式的秒( 00,01,
- 读《研磨设计模式》-代码笔记-组合模式
bylijinnan
java设计模式
声明: 本文只为方便我个人查阅和理解,详细的分析以及源代码请移步 原作者的博客http://chjavach.iteye.com/
import java.util.ArrayList;
import java.util.List;
abstract class Component {
public abstract void printStruct(Str
- 4_JAVA+Oracle面试题(有答案)
chenke
oracle
基础测试题
卷面上不能出现任何的涂写文字,所有的答案要求写在答题纸上,考卷不得带走。
选择题
1、 What will happen when you attempt to compile and run the following code? (3)
public class Static {
static {
int x = 5; // 在static内有效
}
st
- 新一代工作流系统设计目标
comsci
工作算法脚本
用户只需要给工作流系统制定若干个需求,流程系统根据需求,并结合事先输入的组织机构和权限结构,调用若干算法,在流程展示版面上面显示出系统自动生成的流程图,然后由用户根据实际情况对该流程图进行微调,直到满意为止,流程在运行过程中,系统和用户可以根据情况对流程进行实时的调整,包括拓扑结构的调整,权限的调整,内置脚本的调整。。。。。
在这个设计中,最难的地方是系统根据什么来生成流
- oracle 行链接与行迁移
daizj
oracle行迁移
表里的一行对于一个数据块太大的情况有二种(一行在一个数据块里放不下)
第一种情况:
INSERT的时候,INSERT时候行的大小就超一个块的大小。Oracle把这行的数据存储在一连串的数据块里(Oracle Stores the data for the row in a chain of data blocks),这种情况称为行链接(Row Chain),一般不可避免(除非使用更大的数据
- [JShop]开源电子商务系统jshop的系统缓存实现
dinguangx
jshop电子商务
前言
jeeshop中通过SystemManager管理了大量的缓存数据,来提升系统的性能,但这些缓存数据全部都是存放于内存中的,无法满足特定场景的数据更新(如集群环境)。JShop对jeeshop的缓存机制进行了扩展,提供CacheProvider来辅助SystemManager管理这些缓存数据,通过CacheProvider,可以把缓存存放在内存,ehcache,redis,memcache
- 初三全学年难记忆单词
dcj3sjt126com
englishword
several 儿子;若干
shelf 架子
knowledge 知识;学问
librarian 图书管理员
abroad 到国外,在国外
surf 冲浪
wave 浪;波浪
twice 两次;两倍
describe 描写;叙述
especially 特别;尤其
attract 吸引
prize 奖品;奖赏
competition 比赛;竞争
event 大事;事件
O
- sphinx实践
dcj3sjt126com
sphinx
安装参考地址:http://briansnelson.com/How_to_install_Sphinx_on_Centos_Server
yum install sphinx
如果失败的话使用下面的方式安装
wget http://sphinxsearch.com/files/sphinx-2.2.9-1.rhel6.x86_64.rpm
yum loca
- JPA之JPQL(三)
frank1234
ormjpaJPQL
1 什么是JPQL
JPQL是Java Persistence Query Language的简称,可以看成是JPA中的HQL, JPQL支持各种复杂查询。
2 检索单个对象
@Test
public void querySingleObject1() {
Query query = em.createQuery("sele
- Remove Duplicates from Sorted Array II
hcx2013
remove
Follow up for "Remove Duplicates":What if duplicates are allowed at most twice?
For example,Given sorted array nums = [1,1,1,2,2,3],
Your function should return length
- Spring4新特性——Groovy Bean定义DSL
jinnianshilongnian
spring 4
Spring4新特性——泛型限定式依赖注入
Spring4新特性——核心容器的其他改进
Spring4新特性——Web开发的增强
Spring4新特性——集成Bean Validation 1.1(JSR-349)到SpringMVC
Spring4新特性——Groovy Bean定义DSL
Spring4新特性——更好的Java泛型操作API
Spring4新
- CentOS安装Mysql5.5
liuxingguome
centos
CentOS下以RPM方式安装MySQL5.5
首先卸载系统自带Mysql:
yum remove mysql mysql-server mysql-libs compat-mysql51
rm -rf /var/lib/mysql
rm /etc/my.cnf
查看是否还有mysql软件:
rpm -qa|grep mysql
去http://dev.mysql.c
- 第14章 工具函数(下)
onestopweb
函数
index.html
<!DOCTYPE html PUBLIC "-//W3C//DTD XHTML 1.0 Transitional//EN" "http://www.w3.org/TR/xhtml1/DTD/xhtml1-transitional.dtd">
<html xmlns="http://www.w3.org/
- POJ 1050
SaraWon
二维数组子矩阵最大和
POJ ACM第1050题的详细描述,请参照
http://acm.pku.edu.cn/JudgeOnline/problem?id=1050
题目意思:
给定包含有正负整型的二维数组,找出所有子矩阵的和的最大值。
如二维数组
0 -2 -7 0
9 2 -6 2
-4 1 -4 1
-1 8 0 -2
中和最大的子矩阵是
9 2
-4 1
-1 8
且最大和是15
- [5]设计模式——单例模式
tsface
java单例设计模式虚拟机
单例模式:保证一个类仅有一个实例,并提供一个访问它的全局访问点
安全的单例模式:
/*
* @(#)Singleton.java 2014-8-1
*
* Copyright 2014 XXXX, Inc. All rights reserved.
*/
package com.fiberhome.singleton;
- Java8全新打造,英语学习supertool
yangshangchuan
javasuperword闭包java8函数式编程
superword是一个Java实现的英文单词分析软件,主要研究英语单词音近形似转化规律、前缀后缀规律、词之间的相似性规律等等。Clean code、Fluent style、Java8 feature: Lambdas, Streams and Functional-style Programming。
升学考试、工作求职、充电提高,都少不了英语的身影,英语对我们来说实在太重要