
scSLAM-seq reveals core features of transcription dynamics in single cells
摘要
单细胞转录组测序(single-cell RNA-seq, scRNA-seq)强调了细胞间表达异质性在健康和疾病表型变异中的重要作用。然而,目前的scRNA-seq方法仅仅提供了基因表达的一个快照,很少传达关于转录的真实时间动态和随机特性的信息。scRNA-seq分析的另一个关键限制是每个细胞的RNA图谱只能分析一次。原文作者开发了单细胞巯基连接的RNA烷基化代谢标记测序技术scSLAM-seq,它整合了代谢RNA标记、生化核苷转化和scRNA-seq,通过区分每个单细胞数千个基因的新老RNA来直接记录转录活性。通过使用scSLAM-seq来研究裂解性巨细胞病毒在单个小鼠成纤维细胞中的感染情况。从旧RNA推断出的细胞周期状态和感染剂量使得能够基于新RNA进行剂量反应分析。因此,scSLAM-seq既可视化又解释了单细胞水平转录活性的差异。此外,scSLAM-seq描述了宿主基因表达中的“开-关”控制和转录动力学,以及与启动子内在特征(BP–TATA-box相互作用和DNA甲基化)相关的广泛基因特异性差异。因此,基因特异性而非细胞特异性的特征解释了单个细胞之间的转录组异质性和转录对扰动的反应。
<更多精彩,可关注微信公众号:AIPuFuBio和大型免费综合生物信息学资源和工具平台AIPuFu:www.aipufu.com>
前言
SLAM-seq技术可将细胞短暂暴露于核苷类似物4-硫尿苷(4sU)。4sU在转录过程中被整合到新的RNA中,并在RNA测序前用碘乙酰胺转化为胞嘧啶类似物。源自新RNA的测序读段(reads)可以在总RNA读段库中根据特征性的铀转化为碳的转化进行鉴定。通过应用SLAM-seq技术在单细胞水平上解决裂解性小鼠巨细胞病毒(CMV)感染的发作。在优化单细胞测序(scSLAM-seq)(图1)后,原文作者在107个小鼠成纤维细胞上构建scSLAM-seq,并使用(大量)SLAM-seq并行分析匹配较大(1
× 10)细胞群体(n = 2)的全局转录变化。在对具有超过2,500个可检测基因的细胞进行质量过滤后,剩余样品(49个巨细胞病毒感染的,45个未感染的细胞)显示了高质量scSLAM-seq文库的所有特征,包括4%至6%之间的U-C转化率。因此,在单细胞水平上,4sU的结合既有效又均匀。
结果和讨论
由于4sU掺入率约为在50-200个核苷酸中有1个,因此所有来源于新RNA的SLAM序列读段中,有高达50%可能不含铀转化成碳。为了克服这个问题,原文作者开发了“使用SLAM-seq技术对新转录的RNA和衰变率进行全局精确分析的方法(GRAND-SLAM)”,其为一种贝叶斯方法,以包括可信区间在内的完全量化的方式计算新RNA与总RNA的比率(new to total RNA,NTR)(图1)。原文作者还开发了GRAND-SLAM 2.0用于并行分析数百个来源于单个细胞的SLAM-seq文库。通过在成对端模式下分析长读段(150个核苷酸),进一步提高了定量的准确性,这使得4sU转化能够有效地与重叠序列中的测序误差区分开。从而获得了每个细胞数千个基因的精确测量值(90%可信区间< 0.2),从而接单细胞转录组测序scRNA-seq的总灵敏度,并与多细胞SLAM-seq 的数据高相关(相关系数R > 0.73)。
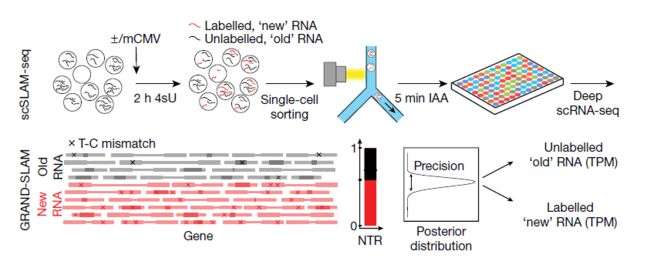
高度可变细胞基因的无偏主成分分析(PCA)结果显示,不能区分感染巨细胞病毒的细胞和未感染的细胞,无论是总RNA还是旧RNA,对新RNA只能有轻微的区分(图2a)。因此,细胞间异质性超过了病毒诱导的变化,这种变化在感染后两小时内很难在总RNA中检测到,这是因为哺乳动物RNA的周转缓慢。相比之下,NTR上的主成分分析可高精度从感染细胞中分离出未感染的细胞(图2a),并显示出与病毒基因表达程度的明显正相关(皮尔逊相关系数R = 0.59,P-value= 7.3 × 10)。
最近的研究表明,从单细胞转录组测序scRNA-seq数据中获得的内含子读段可以用来估计个体细胞中基因表达的时间导数,称为“RNA速度”。这些表明了单个细胞的未来轨迹可投射到基因表达的低维空间中。然而,受感染的细胞不能通过无偏主成分分析从未受感染的细胞中分离出来,该主成分分析是根据相应的RNA速度计算的,或根据使用速度预测未来的表达谱计算的,或直接根据内含子/外显子比率计算的。为了直接将scSLAM-seq与针对更大细胞群计算的RNA速度进行比较,原文作者在数百个未感染(n = 793)和巨细胞病毒感染(n = 353)的细胞上使用相同的实验条件进行了10x Genomics Chromium droplet-based 的单细胞转录组测序。尽管成熟转录物(仅外显子读取)上的主成分分析不能分离未感染和感染的细胞,但使用内含子/外显子比率可以进行区分。然而,在scSLAM-seq和10x数据中没有观察到有意义的RNA速率(图2b)。原文作者使用scSLAM-seq获得的新的和总的RNA水平来代替内含子和外显子的read水平,并确定“NTR速度”。值得注意的是,这些进一步区分了感染细胞和未感染细胞(图2c)。
为了直接比较NTRs和RNA的速度,原文作者问进一步分析了哪一个能最好地预测一个基因在大细胞群中是上调还是下调。虽然这在某种程度上是可能的,分别使用从scSLAM-seq或10x数据中的数十或数百个细胞计算的RNA速度(AUC值分别为0.68和0.74),但是通过使用NTRs (AUC > 0.94)可获得更好的结果(图2d)。此外,NTRs相对于velocities (分别来自scSLAM-seq和10x数据,n =131和n = 295),可以确定更多受调控的基因(n =583)。因此,NTR和RNA速率传递了不同但互补的信息,并可以合并到NTR速度中,以更可靠地预测细胞的未来状态。
病毒学中的一个基本问题是为什么一个细胞的感染会导致广泛的裂解性病毒复制而感染的第二个细胞没有。在感染2小时后,大多数细胞的巨细胞病毒感染已经从“立即早期”(仅限于少数基因)发展到“早期”感染阶段,其中大多数病毒基因已经转录。尽管所有细胞中的大多数病毒转录物都是新的,但原文作者也观察到一些病毒基因的大量旧RNA。这代表病毒粒子相关的RNA,由传入的病毒粒子传递到受感染的细胞。它与从病毒源中分离的病毒颗粒相关RNA有很好的相关性(相关系数R =0.48),因此,提供了每个细胞接受的感染剂量的替代标记。相比之下,新的病毒RNA反映了感染的效率。感染剂量解释了52%的感染效果差异。所以,几乎不表达任何新病毒RNA的细胞中的旧病毒RNA的量比其它细胞中的低得多,这表明这些细胞对巨细胞病毒感染的许可性并没有降低,而是接受了低得多的病毒剂量。基于细胞周期特征基因,可以分别从旧的和总的RNA推断4sU代谢标记开始和结束时的细胞周期状态,从而提供细胞周期轨迹(图2e)。虽然裂解性感染始于所有细胞周期阶段,但在G1期感染的细胞导致明显更强的病毒基因表达和细胞周期破坏(P-value <0.05)。这使得新病毒基因表达的解释差异增加到59%(图2f)。因此,在成纤维细胞中单个细胞水平启动裂解性病毒基因表达的效率可以通过感染剂量和细胞周期的相互作用得到很好的解释。
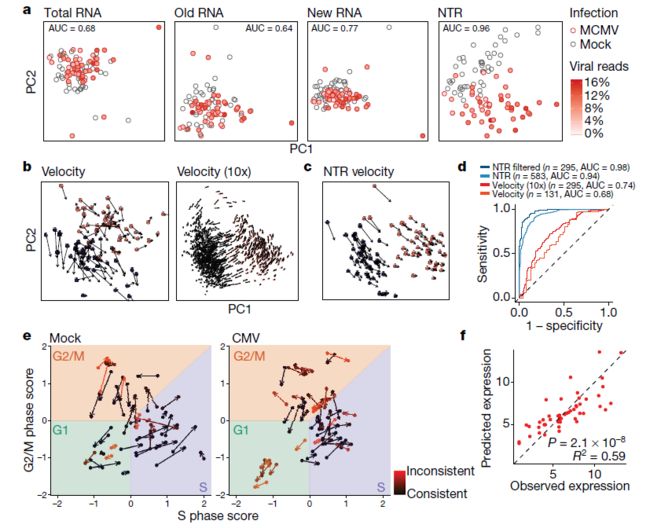
为了评估巨细胞病毒感染对细胞基因表达的影响,原文作者使用单细胞两相检测程序(SC2P)从总的、新的和旧的RNA中鉴定了巨细胞病毒感染和未感染的单细胞之间差异表达的基因。大多数下调(87个基因中的61个)和上调(309个基因中的188个)基因(超过60%)只有通过专门考虑新的RNA才能被发现(图3a)。基因表达的双峰性是单细胞中一个很好描述的特征。双峰性表达的基因在一个细胞亚群中检测不到表达,但可在其他细胞群中检测到表达。scSLAM-seq直接观察细胞中给定基因的启动子在研究的时间范围内是否“开启”。值得注意的是,原文作者发现大多数巨细胞病毒诱导的新RNA的变化更多地与开关动力学相一致,而不是与上调或下调相一致(图3b,c)。
巨细胞病毒感染在感染的前两个小时诱导强烈的I型干扰素和NF-κB反应。根据预测的转录因子靶标和基因本体条目(Gene Ontology terms)进行的基因富集分析表明,干扰素和NF-κB的激活高度依赖于病毒剂量。然而,虽然干扰素的激活只限于大约一半的感染细胞,但在巨细胞病毒感染的大多数细胞都发生了NF-κB激活 (图3d)。病毒剂量依赖性激活所有细胞中的NF-κB与M45被膜蛋白介导的IKK激酶复合体处或上游的NF-κB激活一致。相比之下,干扰素反应需要检测病原体相关的分子模式,受自分泌和旁分泌信号的影响,因此可能在单个细胞之间表现出更大的可变性。为了进行更集中的分析,原文作者根据以前发表的关于NF-κB诱导的数据和干扰素治疗的bulk SLAM-seq数据,定义了巨细胞病毒感染特异的NF-κB和干扰素应答基因组。干扰素(图3e)和NF-κB(图3f)反应的大小在单个细胞之间明显不同,但两者都与病毒基因表达高度相关(斯皮尔曼相关系数ρ > 0.52,P-value < 3 × 10)。因此,大多数NF-κB-和干扰素诱导型基因表达出现在最强感染的细胞中,在S期感染的细胞中诱导最弱的反应。
单细胞水平上的转录活性不是一个连续的过程,而是由间歇的转录爆发组成,中间相隔几分钟到几小时的转录不活跃,表明启动子暂时不允许。原文作者推断,scSLAM-seq应该可以检测到基因的这种爆发因子。为了全面评估转录爆发动力学,原文作者将基因爆发评分(B-评分)定义为所有未感染细胞的正常参考时间分布的标准偏差,其中相应基因的RNA可以可靠地量化(正常参考时间的90%可信区间< 0.2;n = 5540)。从两个独立的生物重复中获得的B-分数高度相关(相关系数R =0.74)。在某些情况下,接近0.5的极端B-分数对应于在每个细胞中仅检测到单个或非常少的mRNA分子(新的或旧的)的基因。
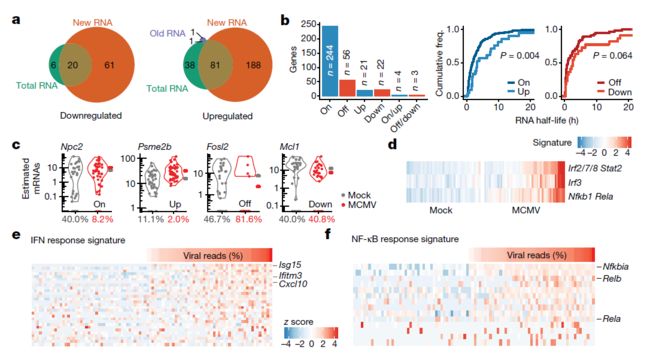
当前的单细胞转录组测序(scRNA-seq)技术要么提供了独特分子的识别标签(UMIs)来估计捕获的mRNA分子的数量,并且是链特异性的,但是仅克隆转录了末端(例如,10x Genomics Chromium单细胞转录组测序),或者可测全长的mRNAs,但是没有UMIs并且失去链特异性(例如,Smart-seq2)。原文作者基于Smart-seq技术开发的scSLAM-seq方法至少部分包含了所有三个特性。4sU的随机合并转化为mRNA分子提供了基于核苷酸转化的独特分子标识(nUMIs),这使得能够估计每个细胞和基因取样的新的mRNA分子数量的下限,并且——通过外推(EnUMIS)——也是旧的mRNA的下限。基于这些对样本多克隆抗体数量的保守估计,发现所有可检测基因中超过30%(5540个中的1718个;矫正后P-value< 0.01,χ检验)的方差(B-得分)大于采样时的预期(图4a)。B分数与表达水平的相关性可以忽略不计。此外,NTR中观察到的异质性不是由细胞周期依赖性差异引起的,而是与mRNA半衰期相关的。

无偏基因本体(Gene Ontoloy)富集分析显示,高B分数的基因与蛋白质磷酸化和泛素化等功能类别相关。启动子分析确定了六个显著富集的低 (TATA box motif)或高 (富含CG和嘌呤motif) B-分数。Correctly placed TATA boxes富集的最明显(P-value< 10-8),与TATA box的启动子一致,在几分钟的时间尺度上频繁的转录爆发。没有观察到与其他核心启动子motif的关联(图4b)。富含CG的motif可以对应于显示振荡激活模式的特定转录因子的结合位点,或者反映相应启动子内富含CpG的区域。超过50%的哺乳动物转录起始于靠近CpG岛的启动子(CGI启动子),这些启动子代表几十到几百个核苷酸的富含CpG的区域。CpG岛的高甲基化是基因沉默的表观遗传控制机制。来自小鼠成纤维细胞的亚硫酸氢盐测序数据揭示了甲基化的降钙素基因相关肽启动子与低B-评分的显著相关性,而甲基化的非降钙素基因相关肽启动子倾向于显示高B-评分(图4c)。对于含有TATA box的启动子也观察到同样的情况(图4d)。通过重复对1718个具有显著B值的基因和前50%最强表达基因的分析,证实了这些结果。虽然不能完全排除这里所用细胞系的转录活性和核型复杂性的等位基因差异导致了一些观察到的效应,但这并不能解释B-评分与启动子内在特征的强烈相关性。原文作者提出了一个模型,其中基因启动子中的DNA甲基化通过在数小时内的时间尺度上暂时抑制基因的启动子而参与短暂的启动子活性调控。最后,B分数与观察到的单个细胞转录组的异质性高度相关(图4e)。因此,基因启动子特异性特征是细胞间异质性的主要原因。
使用4sU的代谢标记适用于所有主要模式生物,包括脊椎动物、昆虫、植物和酵母。嘌呤类似物6-硫代鸟嘌呤(6sG)现在也能通过氧化-亲核-芳香取代(TimeLapse-seq chemistry)实现从G到A的转换。短而连续的4sU和6sG脉冲,结合巯基-(SH)介导的核苷转化,可以实现单细胞转录活性的两个独立记录。最后,scSLAM-seq与基于CRISPR的扰动相结合,将可极大地提高各种方法破译分子机制的灵敏度,这对发育生物学、感染和癌症具有重要意义。
<更多精彩,可关注微信公众号:AIPuFuBio和大型免费综合生物信息学资源和工具平台AIPuFu:www.aipufu.com>