RNAseq实际操作(实战)
首先声明,虽然是实战,但是其实是学习笔记而已,初学,参考了大量大神的博客和帖子,还有大神引用的大牛的帖子,参考列表见最后。
1、软件安装
1.1 硬件系统情况
系统:BioLinux8(ubuntu14.04.2-x64)
吐槽个一下这个系统,普通帐户竟然环境变量错误(source ~/.bashrc),对我等小白来说实在头大,只有用root用户运行了。
后来通过百度错误问题发现,这个系统用的是zsh,所以更新环境变量的代码是 source ~/.zshrc 这样就可以了。
电脑配置:
Lenovo-ThinkPad-E431
CPU系列 英特尔 酷睿i3 3代系列
CPU型号 Intel 酷睿i3 3120M
CPU主频 2.5GHz
总线规格 DMI 5 GT/s
三级缓存 3MB
核心架构 Ivy Bridge
核心/线程数 双核心/四线程
制程工艺 22nm
内存容量 12GB(4GB×1 + 8GB×1)
1.2 软件安装
有简单的不必去重新编译代码了,bioconda几个命令解决,可能软件依赖会有点问题。
1)Miniconda安装
wget https://repo.continuum.io/miniconda/Miniconda3-latest-Linux-x86_64.sh
bash Miniconda3-latest-Linux-x86_64.sh
source ~/.zshrc
一路Enter搞定
2)sratoolkit
conda install -c jfear sratoolkit
3)fastqc
conda install fastqc
发现fastqc是BioLinux自带的,执行这个命令只是升级了java
4)hisat2
conda install hisat2
hisat2 -h #测试
这有个小插曲,提示hisat2依赖于python3.5, 而我装的是3.6,于是百度得知用conda安装3.5版本的:
conda install python=3.5
5)samtools
conda install samtools
samtools
''#samtools也是自带的,执行这个命令会说依赖冲突,因为我的conda是Python3.6版,有点新,不兼容正常。
6)htseq-count
conda install -c bioconda htseq
’#测试
python
>>> import HTSeq
7)R
BioLinux自带了,升级一下到3.4.1
sudo add-apt-repository ppa:marutter/rrutter
sudo apt-get update
sudo apt-get install r-base r-base-dev
8)rstudio
conda install rstudio
rstudio
2、读文章拿到测序数据
直接拿jimmy的脚本修改得来的。仔细分析项目数据后,得到要下载的数据是SRR3589958-62
这个脚本包含了下载数据,sra格式转化为fastq,fastqc对转化的数据进行分析,代码如下:
for ((i=958;i<=958;i++)) ;do wget ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByStudy/sra/SRP/SRP075/SRP075747/SRR3589$i/SRR3589$i.sra;done
ls *sra |while read id; do fastq-dump --gzip --split-3 $id;done
看到大神用的.gz格式才发现太省空间了,至少省了一半以上,膜拜!
3、了解fastq测序数据(fastqc质量控制学习)
1.fastqc
zcat SRR3589956_1.fastq.gz | fastqc -t 4 stdin
fastqc SRR3589956_1.fastq.gz
#t 线程,每个250M内存
2.multiQC
conda install multiqc
# 先获取QC结果
ls *gz | while read id; do fastqc -t 4 $id; done
# multiqc
multiqc *fastqc.zip --pdf
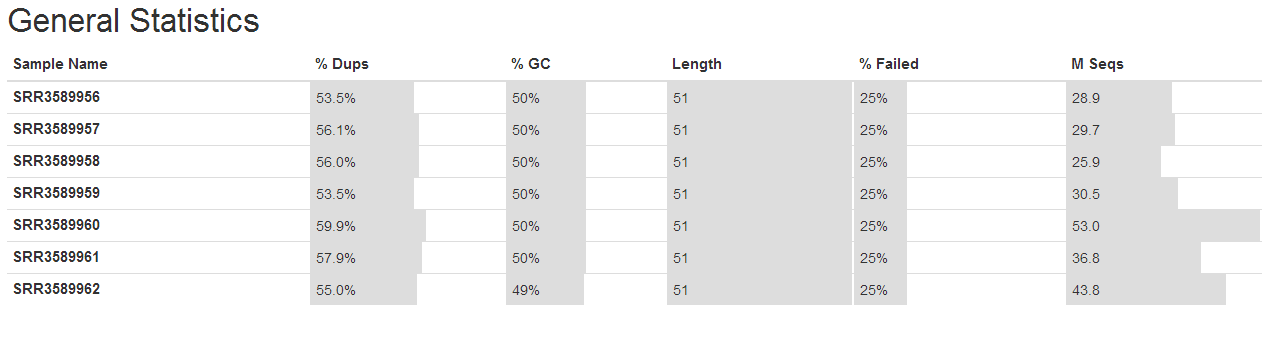
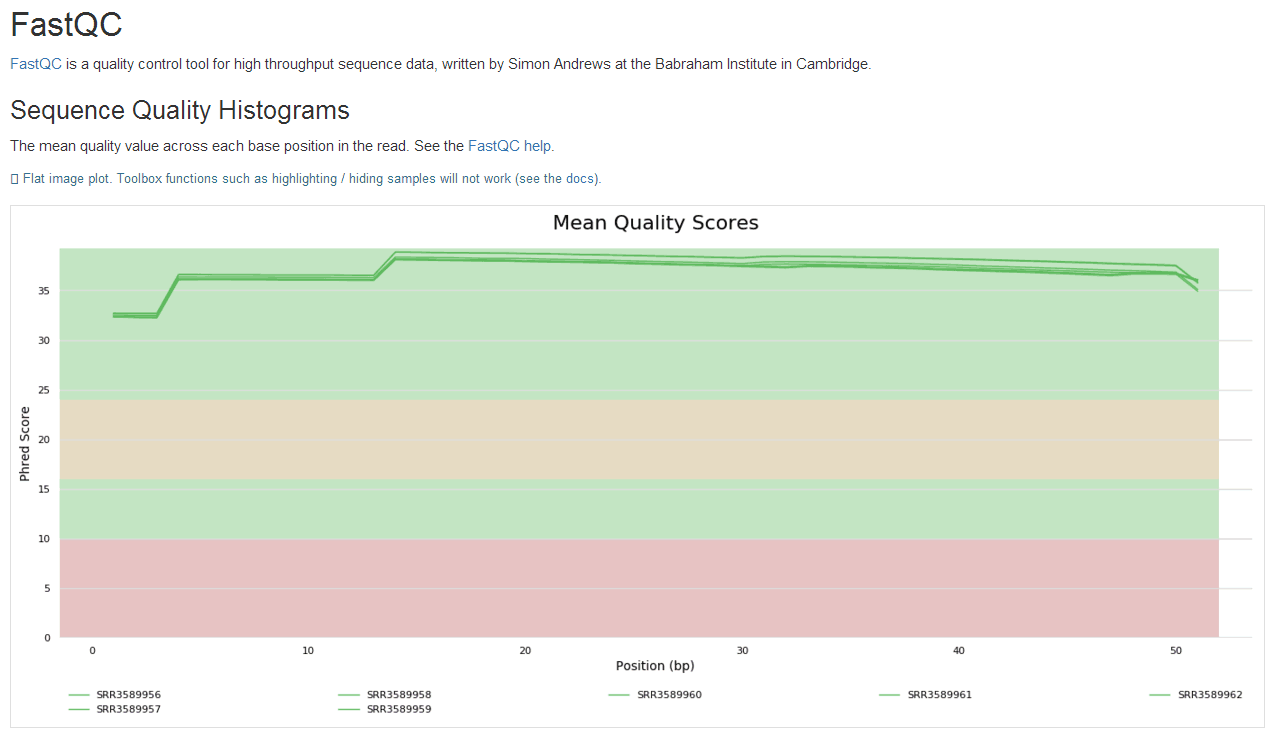
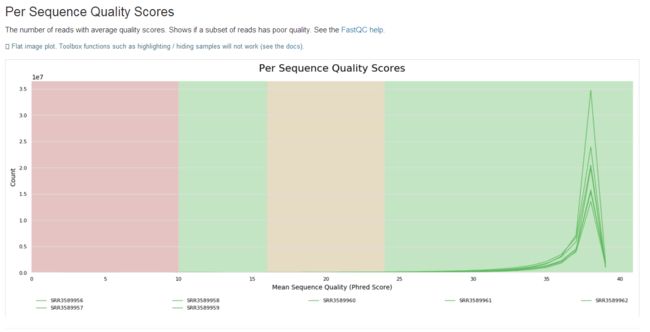

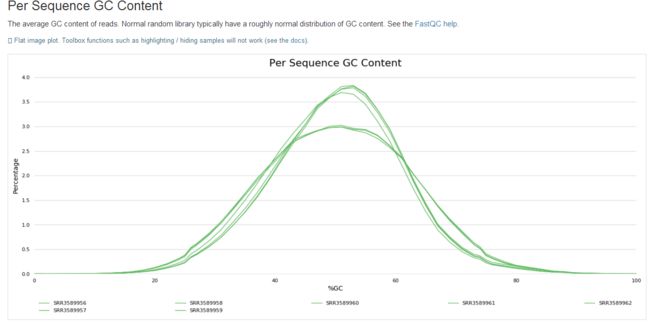
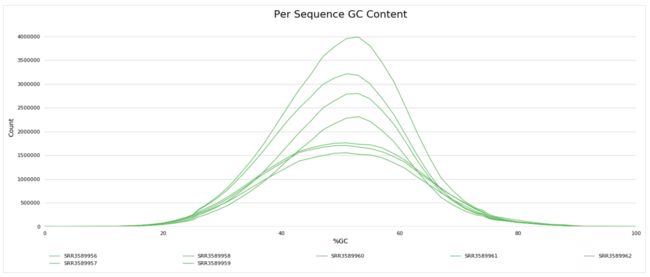
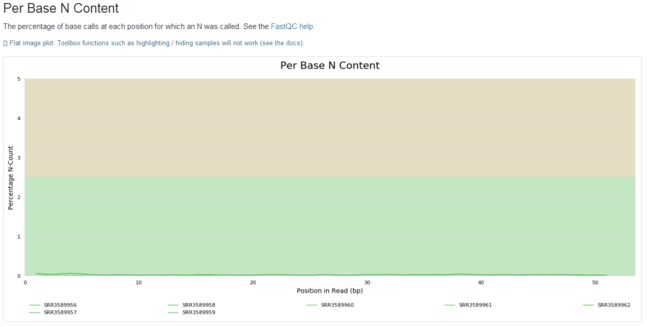
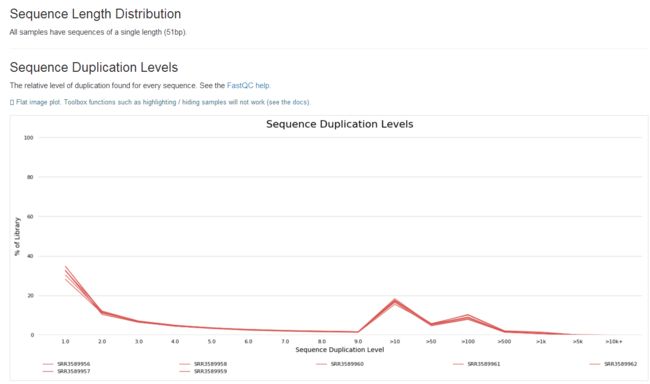
学习笔记是纸质版拍照的,哈哈。



4、了解参考基因组及基因注释
1)下载参考基因组
wget http://hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/chromFa.tar.gz #wget 下载或者其他工具下载
tar -zvf chromFa.tar.gz
cat *.fa > hg19.fa
rm chr*
2)下载基因组注释gtf文件
GTF和GFF之间的区别:
数据结构:都是由9列构成,分别是reference sequence name; annotation source; feature type; start coordinate; end coordinate; score; strand; frame; attributes.前8列都是相同的,第9列不同。
GFF第9列:都是以键值对的形式,键值之间用“=”连接,不同属性之间用“;”分隔,都是以ID这个属性开始。下图中有两个ID,说明是不同的序列。
GTF第9列:同样以键值对的形式,键值之间是以空格区分,值用双引号括起来;不同属性之间用“;”分隔;开头必须是gene_id, transcipt_id两个属性。
wget ftp://ftp.sanger.ac.uk/pub/gencode/Gencode_human/release_26/GRCh37_mapping/gencode.v26lift37.annotation.gtf.gz]ftp://ftp.sanger.ac.uk/pub/genco ... 7.annotation.gtf.gz
3)下载IGV(Integrative Genomics Viewer)
下载地址为: http://software.broadinstitute.org/software/igv/download
4)genome -> Load Genome From Files加载之前得到基因组文件
5)Tool -> Run igvtools,进行排序
6)加载gff基因注释文件,File -> Load From Files
7)可视化分析
同样推荐阅读生信宝典公众号文章。
https://mp.weixin.qq.com/s/Q7pqycmQH58xU6hw_LECWA
5、序列比对
5.1 下载index
cd referece && mkdir index && cd index
wget ftp://ftp.ccb.jhu.edu/pub/infphilo/hisat2/data/hg19.tar.gz
tar -zxvf hg19.tar.gz
5.2 比对
mkdir -p RNA-Seq/aligned
for i in seq ‘56 58’
do
hisat2 -t -x reference/index/hg19/genome -1 RNA-Seq/SRR35899${i}_1.fastq.gz -2 SRR35899${i}_2.fastq.gz -S RNA-Seq/aligned/SRR35899${i}.sam
done
5.3 HISAT2输出结果
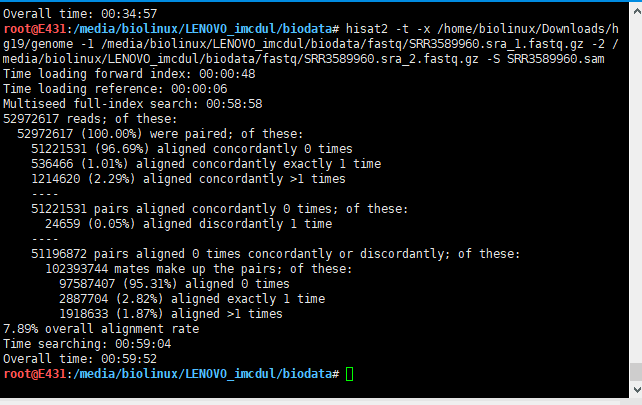
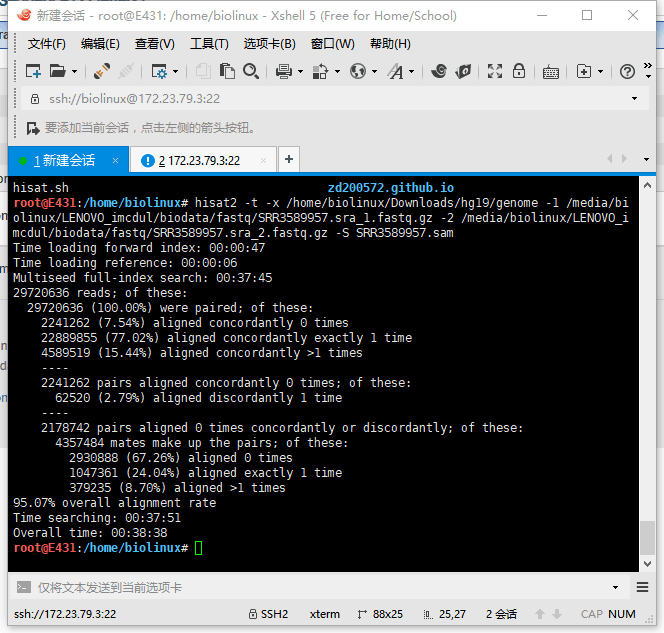
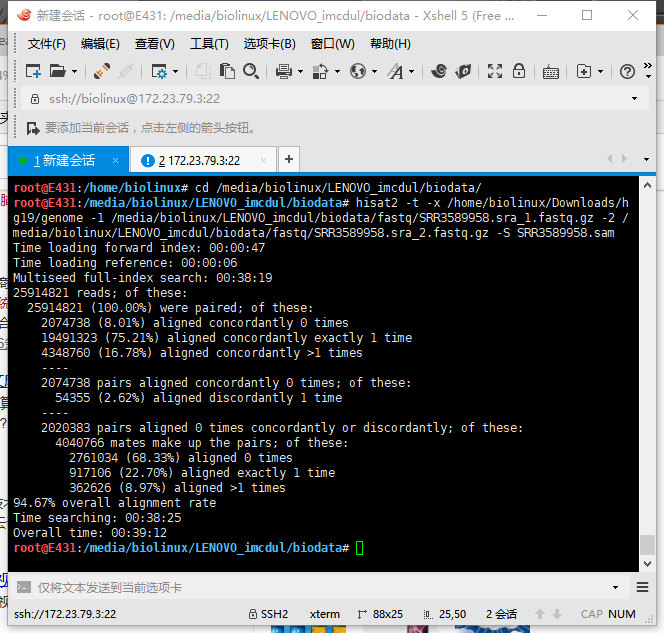
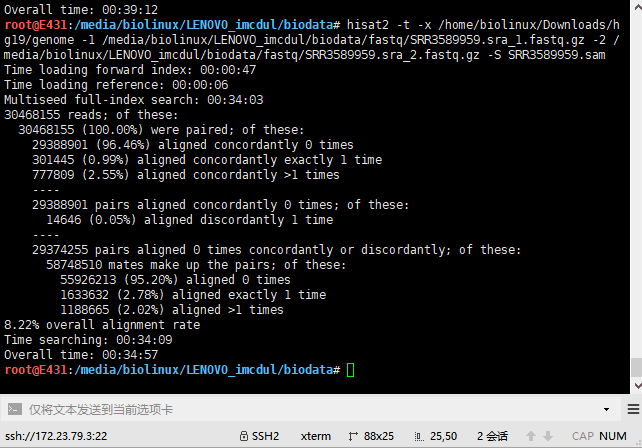
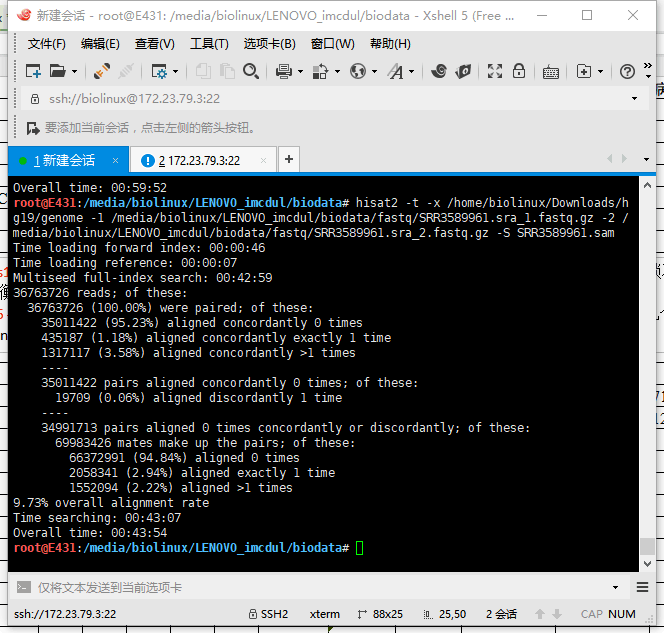

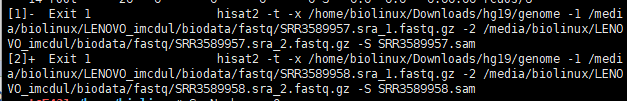
两个同时跑,资源几乎占满。报错,以为硬件不足,后来才发现是硬盘爆了,于是上移动硬盘。
5.4 格式转换,排序,索引
for i in `seq 56 58`
do
samtools view -S SRR35899${i}.sam -b > SRR35899${i}.bam
samtools sort SRR35899${i}.bam SRR35899${i}_sorted.bam
#这里我用的是0.1.19版本,不用加参数 -o
#ps:我加了-o之后重定向输出结果有5个G之工巨,xshell直接死机,直接运行,电脑终端一直不停跳乱码的东西。
samtools index SRR35899${i}_sorted.bam
done
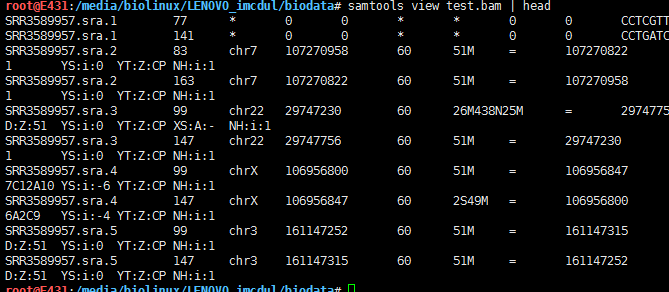
判断sam排序两种方式的不同:
head -100 SRR3589957.sam > test.sam
samtools view -b test.sam > test.bam
samtools view test.bam | head
默认排序
samtools sort test.bam default
samtools view default.bam | head
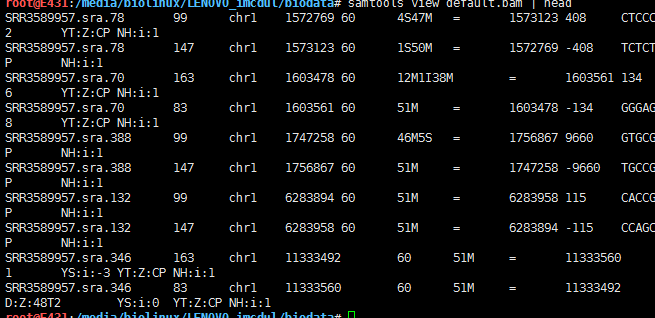
Sort alignments by leftmost coordinates, or by read name when -n is used
samtools sort -n test.bam sort_left
samtools view sort_left.bam | head
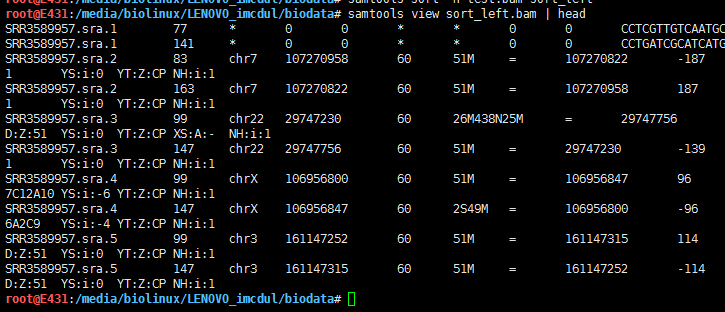
5.5 samtools view
提取1号染色体1234-123456区域的比对read
samtools view SRR3589957_sorted.bam chr1:1234-123456 | head
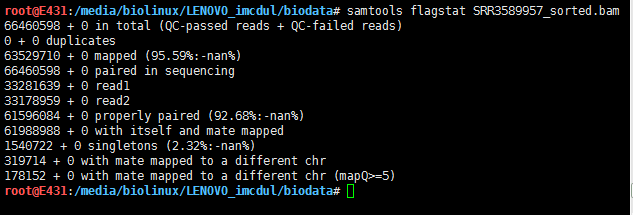
flagstat看下总体情况
samtools flagstat SRR3589957_sorted.bam
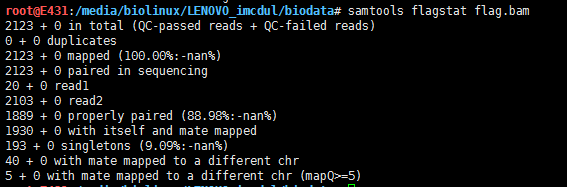
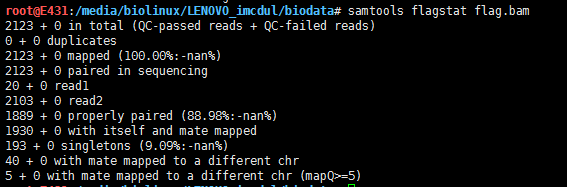
用samtools筛选恰好配对的read,就需要用0x10
samtools view -b -f 0x10 SRR3589957_sorted.bam chr1:1234-123456 > flag.bam
samtools flagstat flag.bam
5.6 比对质控(QC)
# Python2.7环境下
pip install RSeQC
对bam文件进行统计分析(70~90的比对率要求)
bam_stat.py -i SRR3589956_sorted.bam
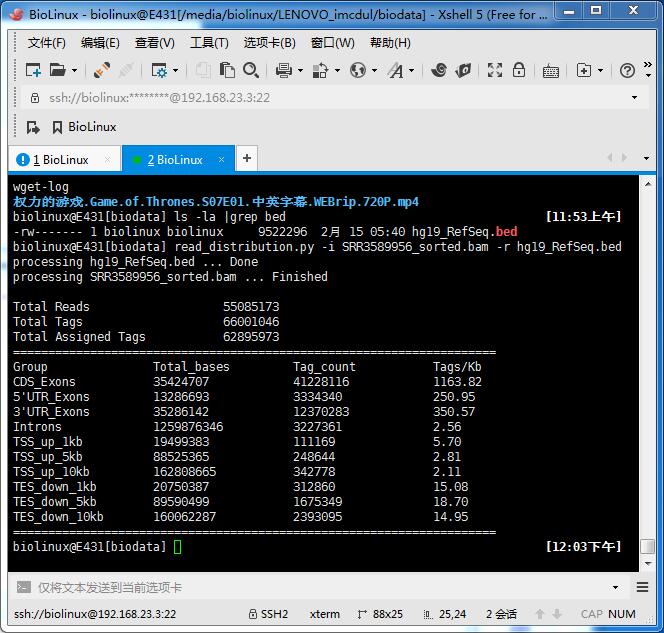
基因组覆盖率的QC
需要提供bed文件,可以直接RSeQC的网站下载(看文件只有1M多,就没有考虑转换):
wget https://downloads.sourceforge.net/project/rseqc/BED/Human_Ho
mo_sapiens/hg19_RefSeq.bed.gz
read_distribution.py -i RNA-Seq/aligned/SRR3589956_sorted.bam -r reference/hg19_RefSeq.bed
5.7 IGV查看
载入参考序列,注释和BAM文件
6 reads计数
# 安装
conda install htseq
# 使用
# htseq-count [options]
htseq-count -r pos -f bam RNA-Seq/aligned/SRR3589957_sorted.bam reference/gencode.v26lift37.annotation.sorted.gtf > SRR3589957.count
循环处理多个BAM文件:
mkdir -p RNA-Seq/matrix/
for i in `seq 56 58`
do
htseq-count -s no -r pos -f bam RNA-Seq/aligned/SRR35899${i}_sorted.bam reference/gencode.v26lift37.annotation.sorted.gtf > RNA-Seq/matrix/SRR35899${i}.count 2> RNA-Seq/matrix/SRR35899${i}.log
done
合并表达矩阵
在生信媛的python脚本上略做了修改
!/usr/bin/python3
def combine_count(count_list):
mydict = {}
for file in count_list:
for line in open(file, 'r'):
#print(line)
if line.startswith('E'):
key,value = line.strip().split('\t')
if key in mydict:
mydict[key] = mydict[key] + '\t' + value
else:
mydict[key] = value
sorted_mydict = sorted(mydict)
out = open('count_out.txt', 'w')
for k in sorted_mydict:
#print(k, mydict[k])
#break
out.write(k + '\t' + mydict[k] +'\n')
count_list = ['SRR3589956.count', 'SRR3589957.count', 'SRR3589958.count']
combine_count(count_list)
简单分析(这里由于对R接触还很少,不少命令不明白,只有运行一下试试)
- 导入数据
options(stringsAsFactors = FALSE)# import data if sample are small
control <- read.table("/media/biolinux/LENOVO_imcdul/biodata/SRR3589956.count",sep="\t", col.names = c("gene_id","control"))
- 数据整合
# merge data and delete the unuseful row
raw_count <- merge(merge(control, rep1, by="gene_id"), rep2, by="gene_id")
raw_count_filt <- raw_count[-1:-5,]
ENSEMBL <- gsub("(.*?)\\.\\d*?_\\d", "\\1", raw_count_filt$gene_id)
row.names(raw_count_filt) <- ENSEMBL
3) 总体情况
summary(raw_count_filt)
4)看几个具体基因
> GAPDH <- raw_count_filt[rownames(raw_count_filt)=="ENSG00000111640",]
#EBI上搜索GAPDH找到ID为ENSG00000111640
> GAPDH
文章研究的AKAP95(ENSG00000105127)的表达量在KD中都降低了
> AKAP95 <- raw_count_filt[rownames(raw_count_filt)=="ENSG00000105127",]
> AKAP95
参考内容列表:
1、http://www.biotrainee.com/forum.php?mod=viewthread&tid=1750#lastpost#jimmy的导航贴
2、转录组(一)](http://www.biotrainee.com/thread-1800-1-1.html) ( HOPTOP )
3、转录组入门(1)](http://www.biotrainee.com/thread-1796-1-1.html)- (青山屋主)
4、转录组入门(1)Mac上软件准备作业
5、PANDA姐的转录组入门(1):计算机资源的准备
6、转录组作业(一):来自零基础的小白
7、转录组入门(2):读文章拿到测序数据
[转录组入门(二) New(HOPTOP)
8、转录组入门(2)-(青山屋主)
9、PANDA姐的转录组入门(2):读文章拿到测序数据
10、转录组入门(3):了解fastq测序数据
11、转录组(三):(HOPTOP)
12、转录组入门(3)-(青山屋主)
13、PANDA姐的转录组入门(3):了解fastq测序数据
转录组入门(4):了解参考基因组及基因注释
14、hoptop的:转录组作业(四)
转录组入门(5): 序列比对
15、转录组入门(5)](http://www.biotrainee.com/thread-1870-1-1.html): 序列比对(HOPTOP)
转录组入门(6): reads计数
16、生信媛
转录组入门(7): 差异基因分析
17、生信媛
18、http://www.bio-info-trainee.com/2218.html
https://jiawen.zd200572.com