前期的到的基因表达量矩阵,可以得到每个基因的表达量,然而由于我们在做实验过程中的重复(包括技术重复与生物学重复)理论上来讲是可以保持表达量在重复中的一致性。因此我们也可通过这个工作来检查我们是否有正确的重复数据。
Trinity工具包提供了一些可以用于检测重复一致性的脚本。我们今天就通过这些脚本进行检查。
在这个工作之前需要两个数据:
1. 基因表达的counts.matrix 文件
2. 生物学重复的表文件
yeyuntian@yeyuntian-rescuer-r720-15ikbn:~/trinitytest/downstr/RSEMout/RSEMout$ l *counts.matrix
RSEM.gene.counts.matrix RSEM.isoform.counts.matrix
yeyuntian@yeyuntian-rescuer-r720-15ikbn:~/trinitytest/downstr/RSEMout/RSEMout$ cat samples.txt
B25 B251
B25 B252
R25 R251
R25 R252
W25 W251
W25 W252
需要注意的是:samples.txt中的名字需要和matrix中的名字一致,否则没办法识别
yeyuntian@yeyuntian-RESCUER-R720-15IKBN:~/Biodata/trinitytest/downstr/RSEMout/RSEMout$ $TRINITY_HOME/Analysis/DifferentialExpression/PtR \ #调用PtR脚本
--matrix RSEM.isoform.counts.matrix \#指定给定的matrix
--samples samples.txt \#样品重复信息
--log2 \#做一个对数处理
--min_rowSums 10 \#过滤数据指标
--compare_replicates #输出的图像参数
为了作为补充,我们获取这个脚本的帮助文件
yeyuntian@yeyuntian-RESCUER-R720-15IKBN:~$ $TRINITY_HOME/Analysis/DifferentialExpression/PtR --help
####################################################################################
#
#######################
# Inputs and Outputs: #
#######################
#
# --matrix matrix.RAW.normalized.FPKM
#
# Optional:
#
# Sample groupings:
#
# --samples tab-delimited text file indicating biological replicate relationships.
# ex.
# cond_A cond_A_rep1
# cond_A cond_A_rep2
# cond_B cond_B_rep1
# cond_B cond_B_rep2
#
# --gene_factors tab-delimited file containing gene-to-factor relationships.
# ex.
# liver_enriched gene1
# heart_enriched gene2
# ...
# (use of this data in plotting is noted for corresponding plotting options)
#
#
# --output prefix for output file (default: "${matrix_file}.heatmap")
#
# --save save R session (as .RData file)
# --no_reuse do not reuse any existing .RData file on initial loading
#
#####################
# Plotting Actions #
#####################
#
# --compare_replicates provide scatter, MA, QQ, and correlation plots to compare replicates.
#
#
#
# --barplot_sum_counts generate a barplot that sums frag counts per replicate across all samples.
#
# --boxplot_log2_dist generate a boxplot showing the log2 dist of counts where counts >= min fpkm
#
# --sample_cor_matrix generate a sample correlation matrix plot
# --sample_cor_scale_limits ex. "-0.2,0.6"
# --sample_cor_sum_gene_factor_expr instead of plotting the correlation value, plot the sum of expr according to gene factor
# requires --gene_factors
#
# --sample_cor_subset_matrix plot the sample correlation matrix, but create a disjoint set for rows,cols.
# The subset of the samples to provide as the columns is provided as parameter.
#
# --gene_cor_matrix generate a gene-level correlation matrix plot
#
# --indiv_gene_cor generate a correlation matrix and heatmaps for '--top_cor_gene_count' to specified genes (comma-delimited list)
# --top_cor_gene_count (requires '--indiv_gene_cor with gene identifier specified')
# --min_gene_cor_val (requires '--indiv_gene_cor with gene identifier specified')
#
# --heatmap genes vs. samples heatmap plot
# --heatmap_scale_limits "" cap scale intensity to low,high (ie. "-5,5")
# --heatmap_colorscheme default is 'purple,black,yellow'
# a popular alternative is 'green,black,red'
# Specify a two-color gradient like so: "black,yellow".
#
# # sample (column) labeling order
# --lexical_column_ordering order samples by column name lexical order.
# --specified_column_ordering comma-delimited list of column names (must match matrix exactly!)
# --order_columns_by_samples_file order the columns in the heatmap according to replicate name ordering in the samples file.
#
# # gene (row) labeling order
# --order_by_gene_factor order the genes by their factor (given --gene_factors)
#
# --gene_heatmaps generate heatmaps for just one or more specified genes
# Requires a comma-delimited list of gene identifiers.
# Plots one heatmap containing all specified genes, then separate heatmaps for each gene.
# if --gene_factors set, will include factor annotations as color panel.
# else if --prin_comp set, will include include principal component color panel.
#
# --prin_comp generate principal components, include top components in heatmap
# --add_prin_comp_heatmaps draw heatmaps for the top features at each end of the prin. comp. axis.
# (requires '--prin_comp')
# --add_top_loadings_pc_heatmap draw a heatmap containing the top feature loadings across all PCs.
# --R_prin_comp_method options: princomp, prcomp (default: prcomp)
#
# --mean_vs_sd expression variability plot. (highlight specific genes by category via --gene_factors )
#
# --var_vs_count_hist create histogram of counts of samples having feature expressed within a given expression bin.
# vartype can be any of 'sd|var|cv|fano'
# --count_hist_num_bins number of bins to distribute counts in the histogram (default: 10)
# --count_hist_max_expr maximum value for the expression histogram (default: max(data))
# --count_hist_convert_percentages convert the histogram counts to percentage values.
#
#
# --per_gene_plots plot each gene as a separate expression plot (barplot or lineplot)
# --per_gene_plot_width default: 2.5
# --per_gene_plot_height default: 2.5
# --per_gene_plots_per_row default: 1
# --per_gene_plots_per_col default: 2
# --per_gene_plots_incl_vioplot include violin plots to show distribution of rep vals
#
########################################################
# Data Filtering, in order of operation below: #########################################################
#
#
## Column filters:
#
# --restrict_samples comma-delimited list of samples to restrict to (comma-delim list)
#
# --top_rows only include the top number of rows in the matrix, as ordered.
#
# --min_colSums min number of fragments, default: 0
#
# --min_expressed_genes minimum number of genes (rows) for a column (replicate) having at least '--min_gene_expr_val'
# --min_gene_expr_val a gene must be at least this value expressed across all samples. (default: 1)
#
#
## Row Filters:
#
# --min_rowSums min number of fragments, default: 0
#
# --gene_grep grep on string to restrict to genes
#
# --min_across_ALL_samples_gene_expr_val a gene must have this minimum expression value across ALL samples to be retained.
#
# --min_across_ANY_samples_gene_expr_val a gene must have at least this expression value across ANY single sample to be retained.
#
# --min_gene_prevalence gene must be found expressed in at least this number of columns
# --min_gene_expr_val a gene must be at least this value expressed across all samples. (default: 1)
#
# --minValAltNA minimum cell value after above transformations, otherwise convert to NA
#
# --top_genes use only the top number of most highly expressed transcripts
#
# --top_variable_genes Restrict to the those genes with highest coeff. of variability across samples (use median of replicates)
#
# --var_gene_method method for ranking top variable genes ( 'coeffvar|anova', default: 'anova' )
# --anova_maxFDR if anova chose, require FDR value <= anova_maxFDR (default: 0.05)
# or
# --anova_maxP if set, over-rides anova_maxQ (default, off, uses --anova_maxQ)
#
# --top_variable_via_stdev_and_mean_expr perform filtering based on the stdev vs. mean expression plot.
# Requires both: (note, if you used --log2 and/or --Zscale, settings below should use those transformed values)
# --min_stdev_expr minimum standard deviation in expression
# --min_mean_expr minimum mean expression value
#
######################################
# Data transformations: #
######################################
#
# --CPM convert to counts per million (uses sum of totals before filtering)
# --CPK convert to counts per thousand
#
# --binary all values > 0 are set to 1. All values < 0 are set to zero.
#
# --log2
#
# --center_rows subtract row mean from each data point. (only used under '--heatmap' )
#
# --Zscale_rows Z-scale the values across the rows (genes)
#
#########################
# Clustering methods: #
#########################
#
# --gene_dist Setting used for --heatmap (samples vs. genes)
# Options: euclidean, gene_cor
# maximum, manhattan, canberra, binary, minkowski
# (default: 'euclidean') Note: if using 'gene_cor', set method using '--gene_cor' below.
#
#
# --sample_dist Setting used for --heatmap (samples vs. genes)
# Options: euclidean, sample_cor
# maximum, manhattan, canberra, binary, minkowski
# (default: 'euclidean') Note: if using 'sample_cor', set method using '--sample_cor' below.
#
#
# --gene_clust ward, single, complete, average, mcquitty, median, centroid, none (default: complete)
# --sample_clust ward, single, complete, average, mcquitty, median, centroid, none (default: complete)
#
# --gene_cor Options: pearson, spearman (default: pearson)
# --sample_cor Options: pearson, spearman (default: pearson)
#
####################
# Image settings: #
####################
#
#
# --imgfmt image type (pdf,svg) with default: pdf
#
# --img_width image width
# --img_height image height
#
################
# Misc. params #
################
#
# --write_intermediate_data_tables writes out the data table after each transformation.
#
# --show_pipeline_flowchart describe order of events and exit.
#
####################################################################################
但是在这个过程中会报错,原因是本地的R包没有安装好,然后回头去安装R包,有些R包在Bioconductor上有些就在CRAN里面。R脚本如下
source("https://bioconductor.org/biocLite.R")
biocLite("Biobase")
installed.packages()
biocLite("qvalue")
help(package='qvalue')
install.packages('fastcluster')
最后结果就是关于一个处理中生物学重复之间的相关性的几个图,放在一个PDF上的
对于图的讲解有机会再讲。(因为我也不知道有什么意义)我先放出来:
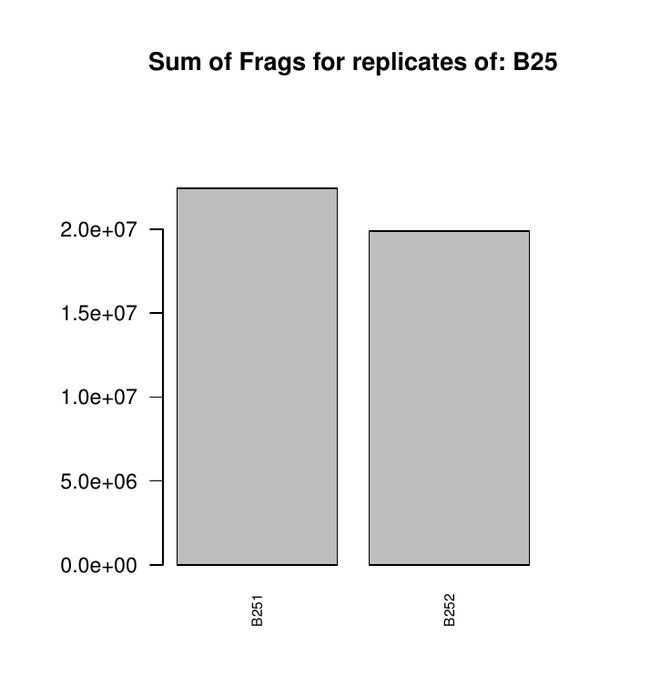
image 1 .png
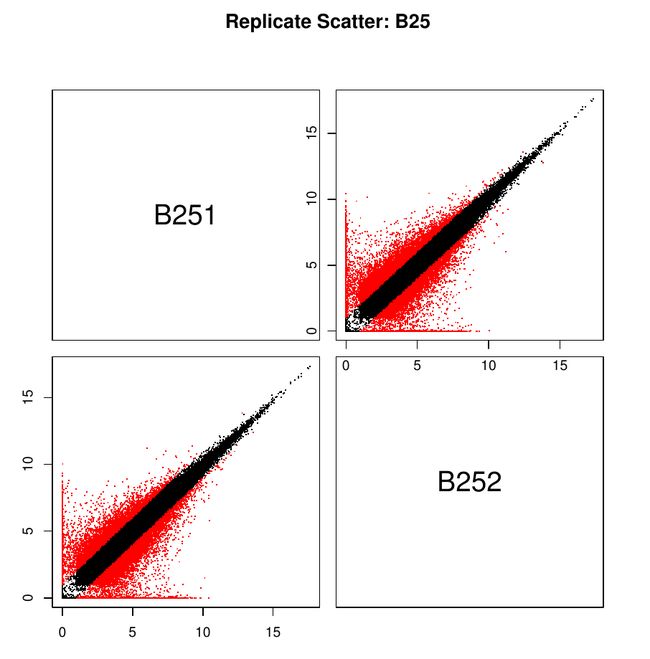
image 2 .png

image 3 .png
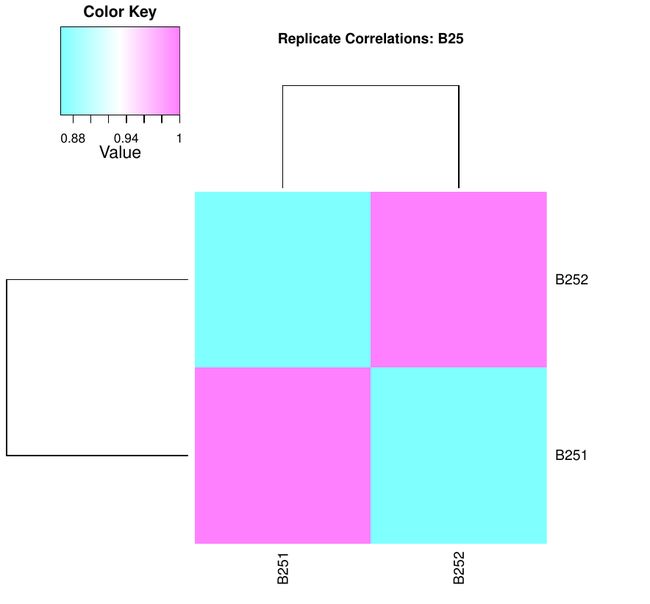
image 4 .png
关于这几张图的解释请大家多多指教,另外我后期通过学习也可以晚上对这个图的解读与分析。
=======================
下面进行跨样本间的相关性检测与作图
$TRINITY_HOME/Analysis/DifferentialExpression/PtR \
--matrix RSEM.isoform.counts.matrix \
--min_rowSums 10 \
--samples samples.txt \ #
--log2 \ #数据转换参数
--CPM \ #数据转换参数
--sample_cor_matrix #输出样品相关性矩阵图
这个代码做出来的结果是不同样本间的数据一致性热图
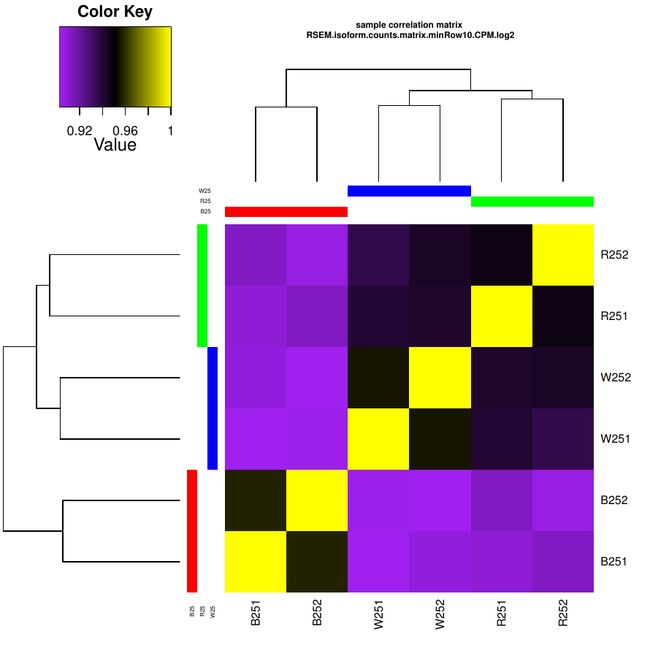
image 5 .png
热图反应处理之间和处理内部的重复之间的一致性
======================
最后一个结果是通过PCA分析对样品重复关系进行检测。
yeyuntian@yeyuntian-RESCUER-R720-15IKBN:~/Biodata/trinitytest/downstr/RSEMout/RSEMout$ $TRINITY_HOME/Analysis/DifferentialExpression/PtR \
--matrix RSEM.isoform.counts.matrix \
--samples samples.txt \
--log2 \
--min_rowSums 10 \
--CPM \
--center_rows \
--prin_comp 3
输出结果为PCA分析图(这个图我也看不懂)
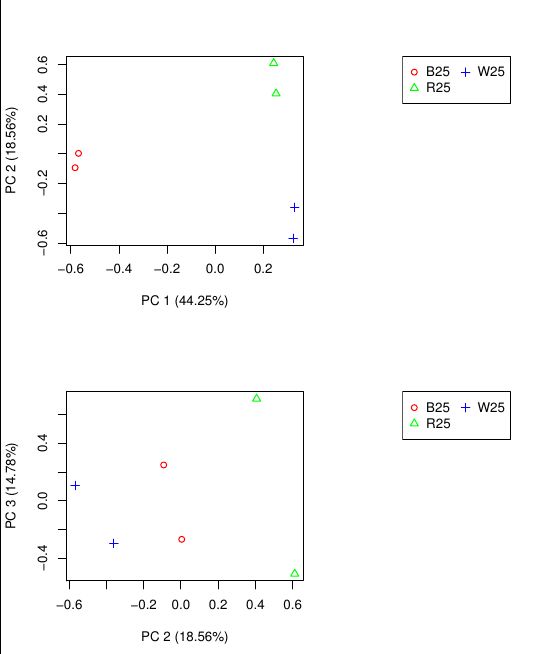
PCA Plot