生信技能树教程

image.png

image.png
image.png

image.png
image.png
这里我要改变顺序,因为目标不一样。我是已经有mRNA,再反过来找miRNA和lncRNA。
三步:
- 靶向TSP1(THBS1)的miRNA预测
- 靶向miRNA的lncRNA的预测
- cytoscape网络构建
靶向TSP1的miRNA
miRTarBase7.0预测
starBase预测
#==============================导入原始数据================
library(data.table)
options(stringsAsFactors = F)
mirTarBase = fread('./dataDownload/hsa_MTI.txt')
starBase = fread('./dataDownload/starBaseV3_hg19_CLIP-seq_miRNA-target_all_THBS1.txt')
save(mirTarBase, mirCode, starBase, file = 'miRNAinput.rdata')
load('./miRNAinput.rdata')
THBS1mirTarBase = subset(mirTarBase, mirTarBase$`Target Gene` == 'THBS1')
if(F){
THBS1miR = starBase[, c(2, 15, 17:21)]
THBS1miR$mirTarBase = ifelse(THBS1miR$miRNAname %in% THBS1mirTarBase$miRNA, 1, 0)
submiRnames = THBS1mirTarBase[!(THBS1mirTarBase$miRNA %in% THBS1miR$miRNAname), 2]
submiR = matrix(rep(0, nrow(submiRnames) * ncol(THBS1miR)), ncol = ncol(THBS1miR),
dimnames = list(submiRnames[[1]], colnames(THBS1miR)))
submiR = as.data.frame(submiR)
submiR$miRNAname = submiRnames[[1]]
submiR$mirTarBase = 1
THBS1miR = rbind(THBS1miR, submiR)
# yige miR
THBS1miR = cbind(THBS1miR[, 1],apply(THBS1miR[, 2:8], 1:2,
function(x)as.numeric(gsub(2, 1, x))))
THBS1miR[, 2:8] %>%
apply(1, sum) -> THBS1miR$Sum
THBS1miR=THBS1miR[order(THBS1miR$miRNAname,THBS1miR$Sum,decreasing = T),]
THBS1miR=THBS1miR[!duplicated(THBS1miR$miRNAname),-9]
}
save(THBS1miR, file ='THBS1miR.rdata')
#==============================画图==============================
install.packages("UpSetR")
library(UpSetR)
data = as.data.frame(THBS1miR[, 2:ncol(THBS1miR)])
library(tidyr)
data %>%
apply(1:2, function(x) gsub(2, 1, x)) %>%
apply(1:2, as.numeric) %>%
as.data.frame() -> data
pdf(file = 'UpSet20.pdf', onefile = F)
upset(data,
sets = colnames(data),#查看特定的几个集合
mb.ratio = c(0.55, 0.45),#控制上方条形图以及下方点图的比例
nintersects = 20,
order.by = "freq", #如何排序,这里freq表示从大到小排序展示
keep.order = F, #keep.order按照sets参数的顺序排序
number.angles = 30, #调整柱形图上数字角度
point.size = 2, line.size = 1, #点和线的大小
mainbar.y.label = "miRNAs Intersections",
sets.x.label = "miRNAs in database",
#坐标轴名称
text.scale = c(1.5, 1.5, 1.5, 1.5, 1.5, 1.5),
queries = list(list(query=intersects,
params=as.list(colnames(data)),
color="red", active=T)))
dev.off()
THBS1miR[, 2:8] %>%
apply(1, sum) -> tmpIndex
targetmiR = THBS1miR$miRNAname[tmpIndex == 7]
save(THBS1miR, data, targetmiR, file = 'mirTarget.rdata')
save(targetmiR, file = 'targetmiR.rdata')
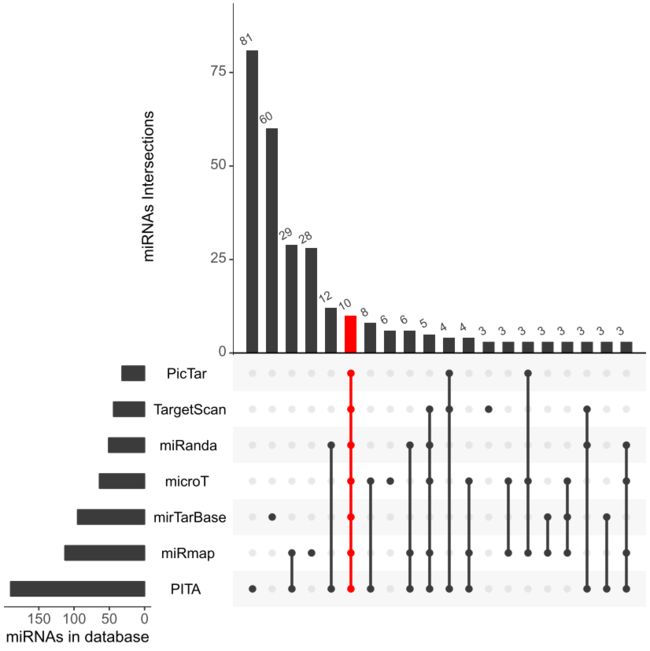
upSet图
miRNA靶向的lncRNA
starBase
miRcode
下载数据starBase:
curl 'http://starbase.sysu.edu.cn/api/miRNATarget/?assembly=hg19&geneType=lncRNA&miRNA=all&clipExpNum=0°raExpNum=0&p
ancancerNum=0&programNum=0&program=None&target=all&cellType=all' > starBaseV3_hg19_CLIP-seq_lncRNA_all.txt &
image.png
image.png
下载miRcode数据:

image.png
rm(list = ls())
lncstarBase = fread('./dataDownload/starBaseV3_hg19_CLIP-seq_lncRNA_all.txt')
miRcode = fread('./dataDownload/mircode_highconsfamilies.txt.gz')
load('./targetmiR.rdata')
lncstarBase = lncstarBase[lncstarBase$miRNAname %in% targetmiR,]
load('./ensem2symbol.Genecode.v22.rdata')
# 根据Ensembl号去除未被GENCODE收录的RNA
ensem2symbol$gene_id %>% substr(1, 15) -> ensem2symbol$gene_id
lncstarBase = lncstarBase[(lncstarBase$geneID %in% ensem2symbol$gene_id),]
# 只保留lncRNA
tmpIndex = miRcode$gene_class %in%
c('lncRNA, intergenic', 'lncRNA, overlapping', 'pseudogene')
lncmiRcode = miRcode[tmpIndex,c(2, 3, 4)]
lncmiRcode = dplyr::distinct(lncmiRcode)#去除重复的行
#miRcode里面,一行的mirna,有多个放一起的情况,需要展开,
#以下代码均是为得到展开后的数据框,代码写的蹩脚,运行很慢,尝试用apply,不行
# 结果保存为'lncRNA-miR-miRcode.rdata'
if(F){
tmpfun =function(x){strsplit(x)[[3]]}
lapply(targetmiR, tmpfun)
gsub('/', '\tmiR-', lncmiRcode$microrna, perl = T) %>%
strsplit(split = '\t') -> tmp
tmpfun = function(i){
if(length(tmp[[i]]) > 1){
tmpRow = data.frame(gene_symbol = unlist(rep(lncmiRcode[i, 1], length(tmp[[i]]))),
gene_class = unlist(rep(lncmiRcode[i, 2], length(tmp[[i]]))),
microrna = tmp[[i]])
return(tmpRow)
}
}
tmpMat = lapply(1:length(tmp), tmpfun)
tmpMat0 = data.table::rbindlist(tmpMat)
#保存结果到lncmiRcode
lncmiRcode = lncmiRcode[!grepl('/', lncmiRcode$microrna),]
lncmiRcode = rbind(lncmiRcode, tmpMat0)
save(lncmiRcode, file = 'lncRNA-miR-miRcode.rdata')
}
load('lncRNA-miR-miRcode.rdata')
#保存结果为'lncRNA-miR-miRcode.rdata'
rm(list=ls(pattern = 'tmp'))#去除临时变量
rm(ensem2symbol, miRcode)
lncstarBase = lncstarBase[, c(2, 4, 5)]
lncstarBase = dplyr::distinct(lncstarBase)
#接着处理lncmiRcode,挑出目标miR的行,变成新的lncmiRcode
targetmiR %>%
lapply(function(x)strsplit(x, split= '-')[[1]][3]) %>%
unlist() %>% gsub('[a-z]', '', .) %>%
unique() -> tmpIndex
lncmiRcode$microrna %>%
lapply(function(x)strsplit(x, split= '-')[[1]][2]) %>%
unlist() %>% gsub('[a-z]', '', .) -> tmpIndexmiRcode
tmpIndexmiRcode %in% tmpIndex -> tmpIndexmiR
lncmiRcode = lncmiRcode[tmpIndexmiR,]
# 对lncmiRcode和lncstarBase取交集
library(dplyr)
lncmiRcode$microrna %>%
lapply(function(x)strsplit(x, split= '-')[[1]]) %>%
lapply(function(x)paste(x[1], x[2], sep = '-')) %>%
unlist() -> tmpIndexmiRcode
tmpIndexmiRcode = gsub(pattern = '[a-z]$', '', tmpIndexmiRcode, perl = T)
lncIntersect = data.frame()
for(i in 1:nrow(lncstarBase)){
lncstarBase$miRNAname[i] %>%
strsplit(., split = '-') %>% unlist() -> tmpmiR
tmpmiR= paste(tmpmiR[2], tmpmiR[3], sep = '-')
tmpmiR = gsub(pattern = '[a-z]$', '', tmpmiR, perl = T)
tmp = filter(lncmiRcode, tmpIndexmiRcode == tmpmiR)
if(lncstarBase$geneName[i] %in% tmp$gene_symbol){
lncIntersect = rbind(lncIntersect, lncstarBase[i, ])
}
}
save(lncIntersect, file = 'lncRNA-miRNA_intersect.rdata')
构建ceRNA网络
#===============================构建ceRNA网络===================================
#得到miR-mRNA, miR-lncRNA后构建合适网络以便导入Cytiscape
rm(list=ls())
load('./lncRNA-miRNA_intersect.rdata')
load('./targetmiR.rdata')
ceRNA = data.frame(miR = targetmiR,
RNA = rep('THBS1', length(targetmiR)),
type = rep('mRNA', length(targetmiR)))
colnames(ceRNA) = colnames(lncIntersect)
ceRNA = rbind(ceRNA, as.data.frame(lncIntersect))
ceRNA$Type = ifelse(ceRNA$geneName == 'THBS1', 'mRNA', 'lncRNA')
fwrite(ceRNA, file = 'ceRNA.csv')
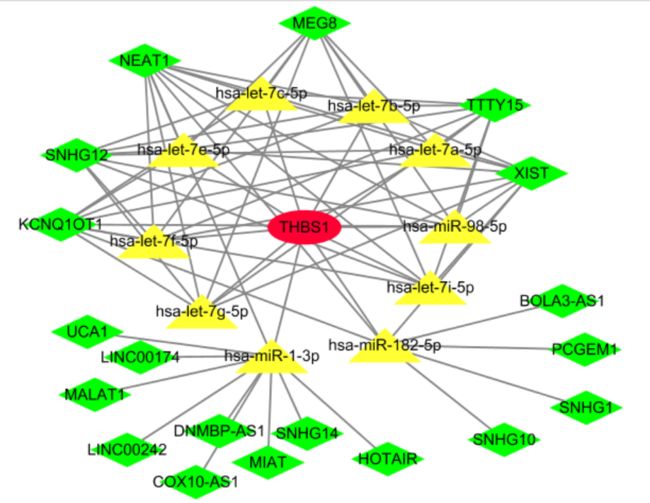
ceRNA network of THBS1