不管在RNA-seq还是ChIP-seq分析中,都涉及到一个概念,就是标准化。
作为一个刚入门的小白,总是听王院长和其他熟悉生信分析的同学们强调标准化的重要性。
为什么说它重要呢?
因为测序读出来的reads落在一个基因内的counts数目取决于基因长度和测序深度。如果一个基因越长,测序深度越高,那么落在其内部的read counts数目就会相对越多。所以在多个样本(不同细胞系条件、不同组织、不同器官、不同个体等等等)中比较某一个基因的表达量,如果不进行数据标准化,比较结果是没有意义的。所以你拿到比对出来的read count是不能直接使用的!(https://www.cnblogs.com/beckygogogo/p/9270698.html)
既然这么重要,所以这个标准化到底是个什么鬼?
简单地说:标准化的对象就是基因长度与测序深度。(https://vip.biotrainee.com/d/63-rpkm-fpkm-rpm-tpm)我在网上搜索了很多关于标准化的教程,这时我才发现学好数学的重要性。。。我是一看到公式就头大啊!!!不管多么简单的公式,都会让我瞬间上头。。。
那么目前有几种标准化的方法?
目前有这么几种标准化方法:RPKM,FPKM,RPM,TPM
First of all:
在使用这几种方法之前,你都需要拿到一个东西:就是read count矩阵。
什么是read count? 答:高通量测序中比对到转录本/基因组上的reads数。(这个在我之前写的几个实战的文章里有提到)
下面就来具体的了解一下几种标准化方法:
(1)RPKM:
Reads Per Kilobase of exon model per Million mapped reads
(每千个碱基的转录每百万映射读取的reads),主要用来对单端测序(single-end RNA-seq)进行定量的方法。
RPKM(软件:Range) 的计算公式:
RPKM= total exon reads/ (mapped reads (Millions) × exon length(KB))
total exon reads:某个样本mapping到特定基因的外显子上的所有的reads。
mapped reads ( Millions ) :某个样本的所有reads总和。
exon length( KB ):某个基因的长度(外显子的长度的总和,以KB为单位)。
优点:tophat-cufflinks流程固定,应用范围广。理论上,可弥补reads Count的缺点,消除样本间和基因间差异。(https://www.jianshu.com/p/c25e84383ae3)

比如现在有3个样品,4个不同的基因,1、2、3样本的总reads数分别是35,45,106。每个基因的read count如下图所示:
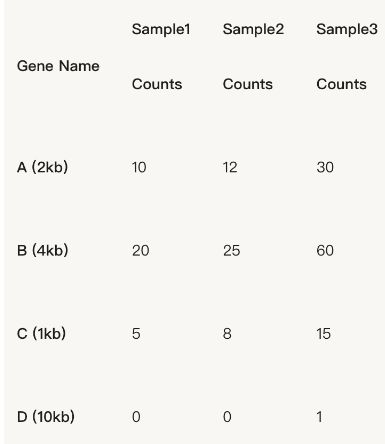
乍一看上去好像read count数差异还挺大的,但是不要高兴的太早。。。我们需要先进行标准化分析。根据公式:RPKM= total exon reads/ (mapped reads (Millions) ×exon length(KB))
我们可以计算每一个样品的RPKM,比如样品一的RPKM=10/(35×2)=1.43,其他的同理 :

现在可以看出,虽然我们拿到的read count值不一样,但是经过标准化以后,这几个基因在3个样品的表达量实际上是差不多的。
(2)FPKM
全称:Fragments Per Kilobase of exon model per Million mapped fragments(每千个碱基的转录每百万映射读取的fragments),主要是针对pair-end测序表达量进行计算。
FPKM (软件:cufflinks)。公式与RPKM一致的。两种计算方法唯一的区别就是:
一个是fragment,一个是read。
如果是PE(Pair-end)测序,每个fragments会有两个reads,FPKM只计算这一对reads能比对到同一个转录本的fragments数量;而RPKM计算的是可以比对到转录本的reads数量,它不管PE的两个reads是否能比对到同一个转录本上。所以,如果是SE(single-end)测序,那么FPKM和RPKM计算的结果将是一致的。(https://www.jianshu.com/p/35e861b76486)
(3)RPM
Reads/Counts of exon model per Million mapped reads (每百万映射读取的reads).
RPM的计算公式:
RPM=(total exon reads×10^6) / mapped reads (Millions)
total exon reads:某个样本mapping到特定基因的外显子上的所有的reads;
mapped reads (Millions) :某个样本的所有reads总和;
优点:利于进行样本间比较。根据比对到基因组上的总reads count,进行标准化。即:不论比对到基因组上的总reads count是多少,都将总reads count标准化为10^6。
缺点:未消除exon长度造成的表达差异,难以进行样本内exon差异表达的比较。
参考文章:关于readsCount、RPKM/FPKM、RPM、TPM的理解
(4)TPM
Transcripts Per Kilobase of exonmodel per Million mapped reads (每千个碱基的转录每百万映射读取的Transcripts),是一种优化后的RPKM,用于同一物种不同组织的比较。(https://zhuanlan.zhihu.com/p/37196518)
TPM (软件:RSEM) 的计算公式:
TPMi={( Ni/Li )*1000000 } / sum( Ni/Li+……..+ Nm/Lm )
Ni:mapping到某个基因上的read数;
Li:某个基因的外显子长度的总和。
在一个样本中一个基因的TPM:先对每个基因的read数用基因的长度进行校正,之后再用校正后的这个基因read数(Ni/Li)与校正后的这个样本的所有read数(sum(Ni/Li+……..+ Nm/Lm))求商。由此可知,TPM概括了基因的长度、表达量和基因数目。TPM可以用于同一物种不同组织间的比较,因为sum值总是唯一的。(https://zhuanlan.zhihu.com/p/37196518)
优点:首先消除exon长度造成的差异,随后消除样本间测序总reads count不同造成的差异。
缺点:因为不是采用比对到基因组上的总reads count,所以特殊情况下不够准确。例如:某突变体对exon造成整体影响时,难以找出差异。
这里有一个例子可以比较容易理解:TPM值就是RPKM的百分比嘛!(转自生信菜鸟团)
这些方法的区别是什么?
FPKM/RPKM:先按列(也就是这个样本的总read数)进行标化,之后再对对个基因的长度进行标准化。
TPM:先对基因长度进行标准化,之后再对列(这个时候就不再是这个样本的总read数了)进行标化。这样使得最终的TPM矩阵的每列都相同(列和都等于1),也就是说每个样本中的TPM的和都是一样的。这样就会使得我们更容易去比较同一个基因在不同样本中所占的read数的比例。而RPKM/FPKM由于最终的表达值矩阵的列和不同,故而不能直接比较同一个基因在不同样本中所占的read数的比例。
参考文章:
RNA-Seq数据标准化方法
哪一种方法适合我的数据分析?
说了那么多,好像每一个看起来都还可以,但是不知道在什么条件下用哪一个方法。这篇文章:RNA-Seq数据标准化方法 是这样说的:
1.学术界已经不再推荐RPKM、FPKM;(但实际上还是有很多人在用的。。。)那么为什么说RPKM和FPKM是错的呢?而又为什么目前很多人都还在用?这里的一篇文章写的非常清楚,非常深奥,写了一大堆公式!!!为什么说FPKM和RPKM都错了? 这篇文章的详细分析过程我没看,真的是心有余而力不足啊。。。但是结论还是看得懂的。
2.比较基因的表达丰度,例如哪个基因在哪个组织里高表达,用TPM做均一化处理;
3.不同组间比较,找差异基因,先得到read counts,然后用DESeq2或edgeR,做均一化和差异基因筛选;如果对比某个基因的KO组和对照,推荐DESeq2。DESeq2和edgeR不用RPKM/FPKM或TPM做均一化,而是直接用原始的read counts做均一化处理。(这里要多说一句:是王院长说的:如果你的RNA-seq没有生物学重复,也就是说每一种处理就一个样品,那就不能用DESeq2来做标准化,要用DESeq做。)
4.如果组间RNA成分差异较大,例如liver跟spleen比,组织特异性表达基因在里面捣乱;再例如常见的某个基因KO组与对照相比,该基因成分在KO组里缺失。就用edgeR/DESeq2作均一化。
下面是我最近在重复一篇文章的分析时遇到的问题:没有生物学重复的RNA-seq怎么分析???
我在之前的RNA-seq分析练习的文章里用到了DESeq2进行read count的标准化,也试过single-end和pair-end的reads分析。但是前两天我在试着重复“Targeting super-enhancer-associated oncogenes in oesophageal squamous cell carcinoma”文章里RNA-seq的部分的时候(这篇文章的ChIP-seq分析部分见https://www.jianshu.com/p/05aae63d7c52),发现这篇文章里的RNA-seq数据没有生物学重复!!!
这个就比较郁闷了,因为我之前用的DESeq2在分析时,在构建矩阵的时候需要输入replicates的read count文件。据王院长说,没有生物学重复的RNA-seq在做标准化的时候,就不能用DESeq2,而是要用DESeq。另外我又在网上查阅了一些资料:
在3个生物学重复不同处理下,如何筛选差异基因
简单使用DESeq2/EdgeR做差异分析
简单使用DESeq做差异分析
readbio/DESeq.R
这些文章都表示:对于没有生物学重复的RNA-seq分析,需要用DESeq标准化。
下面是我的代码:
#导入表达矩阵
>mycounts <- read.csv("/media/yanfang/FYWD/RNA_seq/matrix/KYSE510_treatment_readcount_unnorm.csv")
>head(mycounts)
>rownames(mycounts)<-mycounts[,1]
>mycounts_withoutx<-mycounts[,-1]
>head(mycounts_withoutx)
#利用DESeq进行标准化
>library(DESeq)
>count<-data.frame(row.names=colnames(mycounts_withoutx),condition = c("KYSE510_0hr","KYSE510_2hr","KYSE510_4hr","KYSE510_6hr","KYSE510_12hr"),libType = c("pair-end","pair-end","pair-end","pair-end","pair-end"));
>condition = count$condition;condition;
>cds = newCountDataSet(mycounts_withoutx,condition);
>cds = estimateSizeFactors(cds);
>sizeFactors(cds);
### This step is telling the DESeq you don't have replicates.
#Note the option sharingMode="fit-only". Remember that the default, sharingMode="maximum", takes care of
#outliers, i.e., genes with dispersion much larger than the fitted values. Without replicates, we cannot catch such
#outliers and so have to disable this functionality.
>cds = estimateDispersions(cds ,method="blind",sharingMode="fit-only")
#提取处理2小时样品与对照样品的标准化后的数据,这里conditionA设置为0小时处理,conditionB设置为2小时处理样品
>res_2h = nbinomTest(cds,"KYSE510_0hr", "KYSE510_2hr")
>write.table(res_2h, "KYSE510_2h_vs_0h.res", append = FALSE, quote = FALSE, sep = "\t")
#提取处理4小时样品与对照样品的标准化后的数据
>THZ1_4h_vs_0h = nbinomTest(cds,"KYSE510_0hr", "KYSE510_4hr")
>write.table(THZ1_4h_vs_0h, "KYSE510_4h_vs_0h.res", append = FALSE, quote = FALSE, sep = "\t")
#提取处理6小时样品与对照样品的标准化后的数据
>THZ1_6h_vs_0h = nbinomTest(cds,"KYSE510_0hr", "KYSE510_6hr")
>write.table(THZ1_6h_vs_0h, "KYSE510_6h_vs_0h.res", append = FALSE, quote = FALSE, sep = "\t")
#提取处理12小时样品与对照样品的标准化后的数据
>THZ1_12h_vs_0h = nbinomTest(cds,"KYSE510_0hr", "KYSE510_12hr")
>write.table(THZ1_12h_vs_0h, "KYSE510_12h_vs_0h.res", append = FALSE, quote = FALSE, sep = "\t")
输出的是csv文件,打开之后是这样的:
> a<- read.table("KYSE510_2h_vs_0h.res",sep="\t",header=T)
> head(a)
id baseMean baseMeanA baseMeanB foldChange log2FoldChange pval padj
1 ENSG00000000003 1403.3666530 1292.5892915 1514.1440 1.1714038 0.2282385 0.8127726 1
2 ENSG00000000005 0.0000000 0.0000000 0.0000 NA NA NA NA
3 ENSG00000000419 1559.1391516 1717.4263314 1400.8520 0.8156693 -0.2939437 0.7601109 1
4 ENSG00000000457 507.8739093 624.6008395 391.1470 0.6262351 -0.6752237 0.4944952 1
5 ENSG00000000460 946.8086288 1018.7049871 874.9123 0.8588475 -0.2195260 0.8212624 1
6 ENSG00000000938 0.4519543 0.9039086 0.0000 0.0000000 -Inf 0.9919796 1
我查了网上对baseMean,baseMeanA和baseMeanB的解释:
(http://seqanswers.com/forums/showthread.php?t=23204)
baseMean mean normalised counts, averaged over all samples from both conditions.
baseMeanA mean normalised counts from condition A.
baseMeanB mean normalised counts from condition B.
以上就是无生物学重复的标准化过程。