前前后后接触了一些基因组和转录组拼接的工作,而且后期还会持续进行。期间遇到了各种各样莫名其妙的坑,也尝试了一些不同的方法和软件,简单做一个阶段性小结。上周的今天更新了原理部分,本篇是实战部分。
拼接大致流程
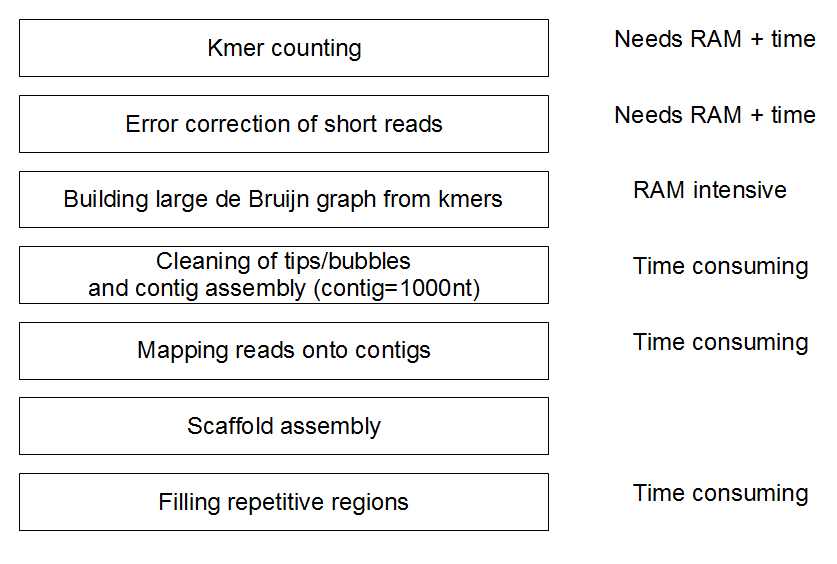
流程的前面4步和DBG算法相关,尤其是一二两步需要较大的内存。拼接结果受 kmer size,kmer coverage cutoff 和 length and coverage parameters 的影响
数据预处理
去接头和低质量reads
类似于通常 RNA-seq 数据处理,略~
使用软件 khmer 进行标准化
digital normalization
关于是否进行 digital normalization 其实一直有不少讨论,最初一篇介绍 digital normalization 的文章指出,所谓digital normalization,是 discards redundant data and both sampling variation and the number of errors present in deep sequencing data sets。其最大的好处是可以降低拼接对内存的要求并且节省时间,而且对于拼出的 contig 没有什么影响。之所以不影响拼接质量,是因为并没有去掉那些低覆盖度的数据。具体可以参考这篇文章 What is digital normalization, anyway。
当然,过了一段时间,还是上文作者又写了一篇博客,说明digital normalization 存在的一些问题。
另附 软件官网使用说明
软件主要功能
- normalizing read coverage ("digital normalization")
- dividing reads into disjoint sets that do not - connect ("partitioning")
- eliminating reads that will not be used by a de - Bruijn graph assembler;
- removing reads with low- or high-abundance k-mers;
- trimming reads of certain kinds of sequencing errors;
- counting k-mers and estimating data set coverage based on k-mer counts;
- running Velvet and calculating assembly statistics;
- converting FASTQ to FASTA;
- converting between paired and interleaved formats for paired FASTQ data
在使用khmer处理数据之前,首先要想清楚是否进行处理。其中最重要的参数是 graph-size filtering 和 graph partitioning。这个软件拼接的时候可以用,计算表达量差异的切忌使用。
另一个是关于 memory 的设置问题,在官方给出的建议中说了一大堆,总的来说就是越大越好 :)
建议使用服务器总内存的一半,如果内存不够的话会报错。一般1 billion 的 mRNAseq 需要-M 16G 16G内存。如果kmer过小,在进行数据清洗的时候,很可能会造成误伤。
Khmer有四种用法
- k-mer counting and abundance filtering
- Partitioning
- Digital normalization
- Read handling: interleaving, splitting, etc.
这里主要使用Digital normalization
关于kmer设置的说明
The interaction between these three parameters and the filtering process is complex and depends on the data set being processed, but higher coverage levels and longer k-mer sizes result in less data being removed. Lower memory allocation increases the rate at which reads are removed due to erroneous estimates of their abundance, but this process is very robust in practice
针对mRANseq的拼接,官方文献给出的建议是
By normalizing to a higher coverage of 20,
removing errors, and only then reducing coverage to 5, digital normalization can retain accurate reads
for most regions. Note that this three-pass protocol is not considerably more computationally expensive
than the single-pass protocol: the first pass discards the majority of data and errors, so later passes are
less time and memory intensive than the first pass
但是针对本身数量不是过大的数据,目前推荐使用single-pass digital normalization的方法。
pair end 数据合并
for i in `ls /projects/zhaofei/wheat_rawdata/LF0*_1.fq.gz`
do
id=`basename $i |sed 's/_1.fq.gz//'`
interleave-reads.py $i /projects/zhaofei/wheat_rawdata/${id}_2.fq.gz -o ${id}_pair.fq.gz --gzip
done
for i in `ls /projects/zhaofei/wheat_rawdata/LF1*_1.fq.gz`
do
id=`basename $i |sed 's/_1.fq.gz//'`
interleave-reads.py $i /projects/zhaofei/wheat_rawdata/${id}_2.fq.gz -o ${id}_pair.fq.gz --gzip
done
single-pass digital normalization
核心步骤,设定相应的cutoff和kmer进行数据处理,cutoff 20; kmer 21/25/27/31/
此处的cutoff 是指:when the median k-mer coverage level is above this number the read is not kept
normalize-by-median.py -p -k 27 -M 50G -C 20 -R LF01.log \
-o LF01k32c28.fq.gz --gzip LF01_pair.fq.gz > runLF01.log 2>&1 &
如果在去街头和低质量数据过程中产生了单端数据,可以在命令中加入-u se.fq 参数
如果想保留生成的dbg文件,可以添加参数 --savegraph normC20k27.ct
去除可能错误的kmer
这一步去除上一步中coverage很高,但是kmer abundanc 低的reads。
filter-abund.py -V -Z 18 normC20k20.ct input.keep.fa && \
rm input.keep.fa normC20k20.ct
# 我自己使用的时候实际没有执行这一步
提取pair end reads
extract-paired-reads.py input.keep.fq
这一步会分别生成仍是pair reads和非 pair reads,生成的数据可以用来后续正式的拼接过程。
分离pair end reads
split-paired-reads.py input.fq.pe
分离后得到的两个fastq文件就可以正式的拼接了。
使用 Trinity 进行拼接
软件介绍
下载地址:https://github.com/trinityrnaseq/trinityrnaseq/wiki
相关文献:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3571712/
Trinity是目前最常用的转录组拼接软件。
拼接过程共分为三步
Inchworm: 拼接过程。从reads到contigs的过程,contigs可能代表一个全长转录本或者一个转录本的一部分。会提取所有的重叠k-mers,根据丰度高低检查每个k-mers,然后将重叠的k-mers延长,形成一个contig
Chrysalis: 将上一步生成的contigs聚类,对每个聚类结果构建DBG图。一个DBG图代表了一个基因的全长转录本。
Butterfly: 根据上一步构建的DBG图和图中的pair end reads 信息寻找最优路径。得到具有可变剪接的全长转录本,同时分开旁系基因的转录本。
直系同源的序列因物种形成(speciation)而被区分开(separated):若一个基因原先存在于某个物种,而该物种分化为了两个物种,那么新物种中的基因是直系同源的;旁系同源的序列因基因复制(gene duplication)而被区分开(separated):若生物体中的某个基因被复制了,那么两个副本序列就是旁系同源的。
常用命令示例
trinityrnaseq-Trinity-v2.4.0/Trinity \
--seqType fq --max_memory 40G --CPU 4 \
--left left.fq.gz \
--right right.fq.gz \
--output test_trinity --full_cleanup \
--no_version_check > test.log 2>&1 &
想要详细了解trinity的处理过程,只需要认真读一下生成的log文件就可以。
大体上是如下几步:
---------------------------
Trinity Phase 1: Clustering of RNA-Seq Reads
---------------------------
In silico Read Normalization
-- (Removing Excess Reads Beyond 50 Coverage --
Jellyfish
-- (building a k-mer catalog from reads) --
Inchworm
-- (Linear contig construction from k-mers) --
Chrysalis
-- (Contig Clustering & de Bruijn Graph Construction) --
------------------------
Trinity Phase 2: Assembling Clusters of Reads
---------------------
Butterfly assemblies are written to \
/projects/zhaofei/wheat_assembly/trinity/LF20_1_trinity.Trinity.fasta
可能出现的报错
需要注意的是,有时候使用trinity拼接一些公用数据会报错
#If your data come from SRA, be sure to dump the fastq file like so:
#SRA_TOOLKIT/fastq-dump --defline-seq '@$sn[_$rn]/$ri' --split-files --gzip file.sra
可以使用命令
fastq-dump --defline-seq '@$sn[_$rn]/$ri' --split-files --gzip input.sra
有的时候不是公用数据仍然报错,是因为pair end 数据第一行@开头的名字不能有空格,必须用1/;2/结尾
#如果中间有空格,结尾正确
zcat ERR037683_1.fastq.gz|awk '{ if (NR%4==1) { print $1"_"$2"" } else { print } }'|gzip >ERR037683_new_1.fastq.gz
#跑循环修改
for i in ERR037679_2.fastq.gz ERR037681_2.fastq.gz ERR037687_2.fastq.gz
do
zcat $i | awk '{ if (NR%4==1) { print $1"_"$2"" } else { print } }'|gzip > ${i/_2.fastq.gz/}_new_2.fastq.gz && rm -f $i
done
#如果结尾不正确
zcat ERR037683_1.fastq.gz|awk '{ if (NR%4==1) { print $1"_"$2"/1" } else { print } }'|gzip >ERR037683_new_1.fastq.gz
#_2.fq 同理替换
有参拼接
首先需要比对得到bam文件,可以使用STAR等软件。如果有多个sample生成多个bam文件,需要在sort之后进行合并
samtools merge my_japonic_j128_sort.bam J1_10_lnc_20160902_q20rmdup.bam \
J2-11-lnc-20160823_q20rmdup.bam J8-10-lnc-20160823_q20rmdup.bam
trinity进行拼接
trinityrnaseq-Trinity-v2.4.0/Trinity --genome_guided_bam my_sort.bam \
--max_memory 60G --SS_lib_type RF --CPU 10 --output tmp_trinity \
--full_cleanup --no_version_check > run.log 2>&1 &
测试小众Bridger拼接软件
github 地址:https://github.com/fmaguire/Bridger_Assembler
文献 https://genomebiology.biomedcentral.com/articles/10.1186/s13059-015-0596-2
按照文章里的这个软件结合了trinity和cufflinks的优点,和trinity相比拼接速度更快占内存更小,可以产生更好的contig(注意:不一定是好事)
安装软件
首先安装boost
a) download latest boost and unpack it.
$ tar zxvf boost_1_47_0.tar.gz
b) change to the boost directory and run ./bootstrap.sh.
$ cd boost_1_47_0
$ ./bootstrap.sh
$ ./b2 install --prefix=
For example,if you want install boost in /home/czheng/local/boost,the commnd is :
$ ./b2 install --prefix=/home/czheng/local/boost
If the boost is installed successfully, you would fild two sub-directories in /home/czheng/local/boost/:
/home/czheng/local/boost/include/
/home/czheng/local/boost/lib/
Note: The default Boost installation directory is /usr/local. Take note of the boost installation
directory, beacuase you need to tell the Bridger installer where to find boost later on.
d) Set the LD_LIBRARY_PATH enviroment variable:
The ~/.bash_profile ($HOME/.bash_profile) or ~/.profile file is executed when you login using console or remotely using ssh.
Append the following command to ~/.bash_profile or ~/.profile file:
$ export LD_LIBRARY_PATH=/home/czheng/local/boost/lib:$LD_LIBRARY_PATH
Save and close the file.
OR
just type the command:
$ export LD_LIBRARY_PATH=/home/czheng/local/boost/lib:$LD_LIBRARY_PATH
Note: please replace "/home/czheng/local/boost/lib" with your own directory "/lib"
If you do not set this variable , you would possible see the follwoing error information:
"error while loading shared libraries: libboost_serialization.so.1.47.0: cannot open shared object file: No such file or dire ctory"
再安装bridger
Building Bridger [Make sure Boost has been installed successfully]
a) Unpack the Bridger and change to the Bridger direcotry.
$ tar zxvf Bridger_r2013-06-02.tar.gz
$ cd Bridger_r2013-06-02
b) Configure Bridger. If Boost is installed somewhere other than /usr/local, you will need to tell
the installer where to find it using --with-boost option.
$ ./configure --with-boost=/home/czheng/local/boost/
Note: please replace "/home/czheng/local/boost/" with your own directory ""
c) Make Bridger.
$ make
note: If you build boost suffessfully without using --prefix option, the following commands may need before "make":
export LIBS="-L/home/czheng/boost_1_47_0/stage/lib" (replace "/home/czheng/boost_1_47_0/" with your own directory)
export CPPFLAGS="-I/home/czheng/boost_1_47_0/"
报错和解决方法
如果安装最新版本的软件,是会报错的。这个问题已经有人在GitHub上提出来了,但是软件的作者已经到阿里巴巴上班了,于是只能自己想办法改一下原始代码。
Splicing Graphs Reconstruction
CMD: ~/software/Bridger_r2014-12-01/src/Assemble --reads both.fa -k 25 --pair_end --fr_strand 2 2>Assemble.log
Error, cmd: ~/software/Bridger_r2014-12-01/src/Assemble --reads both.fa -k 25 --pair_end --fr_strand 2 2>Assemble.log died with ret 256 !
Assemble.log:
[Error] Cannot create directory ./RawGraphs/ !
出现报错以后,如果查看目录会发现已经有/RawGraphs/目录,尝试更改相关源代码解决问题。比如增加生成文件夹的一步。
修改Bridger.pl 文件
## Assemble step:
print "\n### Splicing Graphs Reconstruction ###\n\n";
my $graphdir = "RawGraphs"; #新加
mkdir "$output_directory/$graphdir"; #新加
my $assemble_cmd = "$SRC_DIR/Assemble --reads $target_fa -k $kmer_length ";
核心命令
perl Bridger_r2014-12-01/Bridger.pl --seqType fq -k 31 --left left.fq --right right.fq --CPU 6 --debug -o output
cd-hit 聚类去冗余
cd-hit 是一个聚类软件,可以对DNA序列或者蛋白质序列进行聚类,其本质应该还是多序列比对。为了让拼出来的scaffold更少一些,可以尝试对拼接结果再次聚类,输出每个聚类结果中最长的序列。
但是实际使用过程中,发现效果也一定会特别明显。
cd-hit-est -i input.fasta -o output-cdhit -T 10 -M 200000
会输出两个结果文件,一个包含序列信息,一个是fasta文件。
评估拼接质量
使用软件 transrate
相关文献 :http://genome.cshlp.org/content/early/2016/06/01/gr.196469.115
官方网站 :http://hibberdlab.com/transrate/index.html
主要有三个功能
- by inspecting the contig sequences
- by mapping reads to the contigs and inspecting the alignments
- by aligning the contigs against proteins or transcripts from a related species and inspecting the alignments
这里我们主要使用前两个功能,如果是有参转录组的拼接,可以尝试使用第三个。但如果是为了查看新的转录本,进行第三项评估也没有太大意义。针对转录组拼接而言,第一步中各种长度的统计结果意义也不大,只有回帖率这个指标是最重要的。通过第二部评估,transrate会返回非常多的有用信息。具体结果解读可以参考网站。需要说明的是,在输出结果中,会直接生成一个good.fasta 文件,本质是在计算转录本表达量时不为0的序列。
这个软件进行第二部计算时,调用了SNAP 进行map,调用了salmon 评估转录本表达量,只调用了salmon quant这一步。
以下是几个不同拼接结果的评估比较
bridger
Contig metrics:
-----------------------------------
n seqs 225718
smallest 201
largest 14513
n bases 185942677
mean len 823.78
n under 200 0
n over 1k 58497
n over 10k 34
n with orf 78022
mean orf percent 64.59
n90 312
n70 779
n50 1403
n30 2148
n10 3671
gc 0.51
bases n 551
proportion n 0.0
Read mapping metrics:
-----------------------------------
fragments 9150682
fragments mapped 3428891
p fragments mapped 0.37
good mappings 2163588
p good mapping 0.24
bad mappings 1265303
potential bridges 0
bases uncovered 68339968
p bases uncovered 0.37
contigs uncovbase 140834
p contigs uncovbase 0.62
contigs uncovered 225718
p contigs uncovered 1.0
contigs lowcovered 225718
p contigs lowcovered 1.0
contigs segmented 20264
p contigs segmented 0.09
TRANSRATE ASSEMBLY SCORE 0.042
-----------------------------------
TRANSRATE OPTIMAL SCORE 0.1069
TRANSRATE OPTIMAL CUTOFF 0.1985
good contigs 142380
p good contigs 0.63
khmer treatment and bridger
Contig metrics:
-----------------------------------
n seqs 226334
smallest 201
largest 15190
n bases 187936204
mean len 830.35
n under 200 0
n over 1k 59287
n over 10k 26
n with orf 79146
mean orf percent 63.92
n90 315
n70 789
n50 1412
n30 2175
n10 3695
gc 0.51
bases n 551
proportion n 0.0
Read mapping metrics:
-----------------------------------
fragments 9150682
fragments mapped 3468749
p fragments mapped 0.38
good mappings 2198825
p good mapping 0.24
bad mappings 1269924
potential bridges 0
bases uncovered 70282113
p bases uncovered 0.37
contigs uncovbase 140875
p contigs uncovbase 0.62
contigs uncovered 226334
p contigs uncovered 1.0
contigs lowcovered 226334
p contigs lowcovered 1.0
contigs segmented 20466
p contigs segmented 0.09
TRANSRATE ASSEMBLY SCORE 0.0424
-----------------------------------
TRANSRATE OPTIMAL SCORE 0.1075
TRANSRATE OPTIMAL CUTOFF 0.2096
good contigs 139955
p good contigs 0.62
和trinity拼接结果对比
Contig metrics:
-----------------------------------
n seqs 353674
smallest 201
largest 15185
n bases 243842135
mean len 689.45
n under 200 0
n over 1k 73010
n over 10k 8
n with orf 118991
mean orf percent 69.1
n90 303
n70 592
n50 976
n30 1461
n10 2392
gc 0.51
bases n 0
proportion n 0.0
Contig metrics done in 60 seconds
Calculating read diagnostics...
Read mapping metrics:
-----------------------------------
fragments 9150682
fragments mapped 4171207
p fragments mapped 0.46
good mappings 2960436
p good mapping 0.32
bad mappings 1210771
potential bridges 0
bases uncovered 100987918
p bases uncovered 0.41
contigs uncovbase 241117
p contigs uncovbase 0.68
contigs uncovered 353674
p contigs uncovered 1.0
contigs lowcovered 353674
p contigs lowcovered 1.0
contigs segmented 40211
p contigs segmented 0.11
Read metrics done in 606 seconds
No reference provided, skipping comparative diagnostics
TRANSRATE ASSEMBLY SCORE 0.0459
-----------------------------------
TRANSRATE OPTIMAL SCORE 0.1516
TRANSRATE OPTIMAL CUTOFF 0.2443
good contigs 197643
p good contigs 0.56
自行检验
可以使用 salmon 或者 kaillsto 进行表达量的快速统计分析。
至此,已经完成了常规的转录组拼接工作,可以进行更多的后续分析。比如基因结构注释等等。
基因结构注释
使用PAPS进行GENE结构注释,一定要提前安装好gmap或者blat中的一个或者全部
首先创建conf.txt文件
cp $PASAHOME/pasa_conf/pasa.CONFIG.template $PASAHOME/pasa_conf/conf.txt
##修改conf.txt以下配置
#MYSQLSERVER=localhost
#MYSQL_RW_USER=mysql
#MYSQL_RW_PASSWORD=1234
#MYSQL_RO_USER=readonly
#MYSQL_RO_PASSWORD=1234
配置具体任务的alignAssembly.config
cp $PASAHOME/pasa_conf/pasa.alignAssembly.Template.txt ./alignAssembly.config
#修改alignAssembly.config 的内容
#MYSQLDB=
用trinity生成的文件进行基因注释;-h查看帮助文档。这一步使用的是适合长序列比对的软件GMAP
/PASApipeline-2.1.0/scripts/Launch_PASA_pipeline.pl -c alignAssembly.config -C -R -g genome.fa -t Trinity.fasta --ALIGNERS gmap --transcribed_is_aligned_orient --stringent_alignment_overlap 30.0 --CPU 5
最后的结果文件是{name} #.pasa_assemblies.bed/gff3/gtf/described.txt和{name}.assemblie.fasta 也就是新的注释信息
番外篇
如果想要找到两个拼接结果之间的差异转录本应该怎么做呢?
既然可以利用kmer去冗余,是否可以分别构建两个不同kmer然后找到品种之间的差异kmer,再利用原始数据和差异kmer提取出相关的reads数据。
这样新构建的拼接输入文件就是各自两个品种之间差异的reads,拼出来的结果自然应该是两者之间具有差异的转录本了。
顺着这个思路,我又找到了另外一科kmer相关的处理工具KMC3。
使用工具KMC3
在使用khmer进行数据处理的过程中仍旧感觉这个软件比较吃内存而且浪费时间。于是又去寻找有没有kmer相关更快更省内存的工具,于是找到了KMC3(counting and manipulating k -mer)
KMC的特点是最大限度使用硬盘而不是缓存,使用下来无论是速度还是占用缓存的情况都非常让人惊喜。
除此之外,使用KMC tools 可以对不同databases的kmers进行各种各样的处理。
使用工具KMC,能否直接提取想要比较的差异kmer,然后从fastq文件中直接提取差异kmer,在进行拼接?
计算kmer
分别使用kmer 25,27,31
for i in 25 27 31
do
kmc -m30 -k${i} -t10 -sf10 -sp10 -sr10 test_pair.fq.gz testpair_kmc${i} kmctest
done
通过下面的统计数据可以发现计算速度是非常之快的。
LF15
***********
Stage 1: 100%
Stage 2: 100%
1st stage: 116.666s
2nd stage: 51.8025s
Total : 168.468s
Tmp size : 10862MB
Stats:
No. of k-mers below min. threshold : 278501366
No. of k-mers above max. threshold : 0
No. of unique k-mers : 499147939
No. of unique counted k-mers : 220646573
Total no. of k-mers : 8915675288
Total no. of reads : 68591734
Total no. of super-k-mers : 1324436899
***********
Stage 1: 100%
Stage 2: 100%
1st stage: 114.303s
2nd stage: 49.2716s
Total : 163.575s
Tmp size : 9533MB
Stats:
No. of k-mers below min. threshold : 318563499
No. of k-mers above max. threshold : 0
No. of unique k-mers : 547641627
No. of unique counted k-mers : 229078128
Total no. of k-mers : 8641277484
Total no. of reads : 68591734
Total no. of super-k-mers : 983222805
***********
Stage 1: 100%
Stage 2: 100%
1st stage: 118.874s
2nd stage: 42.6233s
Total : 161.497s
Tmp size : 9026MB
Stats:
No. of k-mers below min. threshold : 336176235
No. of k-mers above max. threshold : 0
No. of unique k-mers : 568550602
No. of unique counted k-mers : 232374367
Total no. of k-mers : 8504082246
Total no. of reads : 68591734
Total no. of super-k-mers : 869491035
LF01
***********
Stage 1: 100%
Stage 2: 100%
1st stage: 120.159s
2nd stage: 51.4074s
Total : 171.566s
Tmp size : 11255MB
Stats:
No. of k-mers below min. threshold : 302154897
No. of k-mers above max. threshold : 0
No. of unique k-mers : 521608251
No. of unique counted k-mers : 219453354
Total no. of k-mers : 9233787753
Total no. of reads : 71035644
Total no. of super-k-mers : 1372680046
***********
Stage 1: 100%
Stage 2: 100%
1st stage: 118.326s
2nd stage: 56.2552s
Total : 174.581s
Tmp size : 9871MB
Stats:
No. of k-mers below min. threshold : 347609349
No. of k-mers above max. threshold : 0
No. of unique k-mers : 576427657
No. of unique counted k-mers : 228818308
Total no. of k-mers : 8949620867
Total no. of reads : 71035644
Total no. of super-k-mers : 1018034018
***********
Stage 1: 100%
Stage 2: 100%
1st stage: 113.98s
2nd stage: 43.8827s
Total : 157.863s
Tmp size : 9346MB
Stats:
No. of k-mers below min. threshold : 367985226
No. of k-mers above max. threshold : 0
No. of unique k-mers : 600562089
No. of unique counted k-mers : 232576863
Total no. of k-mers : 8807539475
Total no. of reads : 71035644
Total no. of super-k-mers : 900199909
计算两组数据kmer子集
kmer25
kmc_tools -t10 -v simple LF01kmc25 LF15kmc25 \
intersect LF01_LF15_kmc25_inter_min -ocmin \
intersect LF01_LF15_kmc25_inter_max -ocmax \
union LF01_LF15_kmc25_union \
counters_subtract LF01_only_diff \
kmers_subtract LF01_only_sub \
reverse_kmers_subtract LF15_only_sub \
reverse_counters_subtract LF15_only_diff \
counters_subtract LF01_only_diff_ci5 -ci5 \
kmers_subtract LF01_only_sub_ci5 -ci5 \
reverse_kmers_subtract LF15_only_sub_ci5 -ci5 \
reverse_counters_subtract LF15_only_diff_ci5 -ci5
根据子集kmer提取对应序列
kmc_tools filter LF15_only_diff_ci5 \
../LF15_pair.fq.gz LF15_pair_only_diff_ci5.fq
提取并分离pair序列
for i in `ls *sub*.fq`
do
echo $i
extract-paired-reads.py $i
split-paired-reads.py *pe && rm *pe *se
done
拼接样本的特有序列
perl Bridger_r2014-12-01/Bridger.pl --seqType fq -k31 \
--left LF01_only_sub_kmc31_ci5_1.fq \
--right LF01_only_sub_kmc31_ci5_2.fq \
--CPU 6 --debug -o LF01_only_sub_kmc31_ci5
评估拼接质量
LF01_pair_only_sub_ci5
transrate --assembly LF01_pair_only_sub_ci5.fasta \
--left ../LF01_pair_only_sub_ci5_1.fq \
--right ../LF01_pair_only_sub_ci5_2.fq \
--threads 20 --output LF01_pair_only_sub_ci5.stats
Contig metrics:
-----------------------------------
n seqs 105761
smallest 201
largest 9487
n bases 49478060
mean len 467.83
n under 200 0
n over 1k 8471
n over 10k 0
n with orf 17698
mean orf percent 70.96
n90 236
n70 334
n50 536
n30 963
n10 2044
gc 0.5
bases n 127
proportion n 0.0
Read mapping metrics:
-----------------------------------
fragments 2730635
fragments mapped 2412155
p fragments mapped 0.88
good mappings 1968236
p good mapping 0.72
bad mappings 443919
potential bridges 9198
bases uncovered 3064853
p bases uncovered 0.06
contigs uncovbase 34327
p contigs uncovbase 0.32
contigs uncovered 3605
p contigs uncovered 0.03
contigs lowcovered 99347
p contigs lowcovered 0.94
contigs segmented 11082
p contigs segmented 0.1
TRANSRATE ASSEMBLY SCORE 0.2736
-----------------------------------
TRANSRATE OPTIMAL SCORE 0.4162
TRANSRATE OPTIMAL CUTOFF 0.0928
good contigs 93367
p good contigs 0.88
LF15_pair_only_sub_ci5
transrate --assembly LF15_pair_only_sub_ci5.fasta \
--left ../LF15_pair_only_sub_ci5_1.fq \
--right ../LF15_pair_only_sub_ci5_2.fq \
--threads 20 --output LF15_pair_only_sub_ci5.stats
Contig metrics:
-----------------------------------
n seqs 131908
smallest 201
largest 8950
n bases 59642308
mean len 452.15
n under 200 0
n over 1k 9144
n over 10k 0
n with orf 23291
mean orf percent 73.01
n90 236
n70 333
n50 508
n30 836
n10 1660
gc 0.53
bases n 305
proportion n 0.0
Read mapping metrics:
-----------------------------------
fragments 2071137
fragments mapped 1759126
p fragments mapped 0.85
good mappings 1378649
p good mapping 0.67
bad mappings 380477
potential bridges 9731
bases uncovered 3974836
p bases uncovered 0.07
contigs uncovbase 45862
p contigs uncovbase 0.35
contigs uncovered 4966
p contigs uncovered 0.04
contigs lowcovered 126884
p contigs lowcovered 0.96
contigs segmented 11948
p contigs segmented 0.09
TRANSRATE ASSEMBLY SCORE 0.2477
-----------------------------------
TRANSRATE OPTIMAL SCORE 0.3932
TRANSRATE OPTIMAL CUTOFF 0.2835
good contigs 103637
p good contigs 0.79
至此,似乎找到了两者之间差异的de novo 转录本,但是效果怎么样,还需要后续分析进一步验证。
利用TransDecoder找ORF
利用TransDecoder将上述两个品种uinq的DNA序列转换为cds和蛋白序列
ransDecoder.LongOrfs -t good.LF15_pair_only_sub_ci5.fasta \
>transdecoder.log 2>&1 &
双向blast
将上述两个蛋白序列进行双向blast
makeblastdb -in good.test.pep -dbtype prot -out good.pep
blastp -query a.pep -db good.pep -num_threads 8 -max_target_seqs 1 -outfmt 6 > a.outfmt6
blastp -query b.pep -db good.pep -num_threads 8 -max_target_seqs 1 -outfmt 6 > b.outfmt6
这样似乎可以继续缩小差异转录本的范围
参考资料
参考资料1
参考资料2
参考资料3
