3.5 Extensions for Reactions
The SMILES language is extended to handle reactions. There are two areas where SMILES is extended: distinguishing component parts of a reactions and atom maps.
Component parts of a reaction are handled by introducing the ">" character as a new separator. Any reaction must have exactly two > characters in it. ">>" is a valid reaction SMILES for an empty reaction. Each of the ">"-separated components of a reaction must be a valid molecule SMILES.
As an aside, molecule SMILES never have a ">" character. In a program, one can quickly determine if a SMILES refers to a reaction or molecule by searching for a ">" character in the string.
Reaction SMILES Grammar:
reaction : reactant '>' agent '>' product
| reactant '>>' product
;
reactant,
agent,
product : molecule
|
;
molecule : SMILES
;
SMILES: a valid molecule specification in the SMILES language.
For example:
C=CCBr>>C=CCI
This is a valid reaction. Note that there are no agent molecules. Also note that several atoms are missing from the reaction (the product "Br" and the reactant "F").
[I-].[Na+].C=CCBr>>[Na+].[Br-].C=CCI
This is a more complete version of the same reaction. It has been canonicalized. It would form the root of a datatree when stored in a THOR database.
C=CCBr.[Na+].[I-]>CC(=O)C>C=CCI.[Na+].[Br-]
This version of the reaction includes an agent. Note that the SMILES does not indicate how the agent participates. Whether the agent is a solvent, catalyst, or performs another function within the reaction must be stored separately as data. This SMILES could be stored in a THOR database as an absolute SMILES and would appear on the same datatree page as the previous example.
In the above example, note that the reaction is ambiguous with respect to the carbon atoms involved. One might assume that a normal Sn2 displacement is occurring. In fact, an equally reasonable allylic displacement is possible, via either an Sn1-like allyl cation. Recognize that the reaction SMILES given above do not say which carbons are which and hence do not discriminate between the two alternate mechanisms.
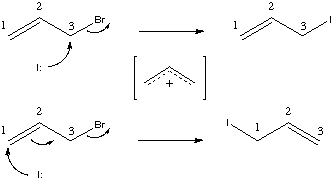
This case demonstrates the use and need for atom maps for reaction processing. Atom maps are used primarily to further define the overall reaction in cases where the reaction mechanism may not be evident from the reactant and product molecules. Atom maps are non-negative integer atom modifiers. They follow the ":" character within an atom expression. They must be the last modifier within the atom expression:
SMILES Atom Expression Grammar:
atom : SYMBOL
| [ WEIGHT SYMBOL mods ]
| [ WEIGHT SYMBOL mods : CLASS ]
;
mods : mod mods
;
mod : HCOUNT | CHARGE | CHIRAL
;
CLASS = non-negative integer class value.
WEIGHT = atomic weight.
SYMBOL = atomic symbol.
HCOUNT = Atom hydrogen count specification.
CHARGE = Atom charge specification.
CHIRAL = Atom chirality specification.
Atom maps are an atomic property. They can legally appear in a SMILES for any atom, whether or not it is part of a reaction. Atom with atom map labels in a molecule SMILES are considered valid; the atom maps are ignored for molecule processing. Absolute and unique SMILES generated by the system for molecules never include atom maps.
Finally, there are some differences in the handling of atom maps and agent components in the unique versus absolute SMILES for reactions. Atom maps and agent components are not part of the unique SMILES specification. This is important for the THOR database, where the datatree roots are formed from the unique SMILES. The net result is that each reaction datatree may contain multiple specific reactions with different agents and atom maps.
3.5.1 Reaction Atom Maps
Atom mappings are properties of the atoms in the reaction molecules. The mappings represent equivalence classes of atoms within a reaction. In effect, the map tells the computer which atoms are the same on the reactant and products sides of a reaction. Without this map information, it is difficult to derive the reaction bond changes which occur.
Within the SMILES language, atom maps are represented as a non-negative numeric atom modifier following the ":" character (e.g. [CH3:2] is a carbon in class 2).
Within the Daylight toolkit, the atom maps are manipulated as sets of mapped atoms. The atom map class numbers which are used in SMILES do not appear in the toolkit interface to a reaction. The map class numbers in SMILES do not have any additional significance, except to associate all atoms with the same map class label to one another.
There are no requirements for completeness or uniqueness of the atom mappings. Atom mappings are independent of the connectivity and properties of the underlying molecules. This is so for several reasons: first, there are limits to the valence representation of molecules which appear when processing reactions. For example the oxygens in sodium acetate (CC(=O)[O-].[Na+]) are chemically indistinguishable, even though the valence model used in the toolkit requires that they be connected differently. Some systems (CAS, for example) recognize this equivalence in their structural representation (the tautomer bond). It is often useful to map these to the same class for reaction purposes: [CH3:1][C:2](=[O:3])[O-:3].[Na+:4]
A second case is where there is ambiguity in a reaction mechanism which one wants to express:

can undergo a cope rearrangement before reaction (which yields the same molecule graph). In effect, there are two distinct mechanisms by which the product is produced. This can be expressed as part of a reaction by: [CH2:1]=[CH:2][CH2:1][CH2:3][C:4](C)[CH2:3]
A third case is simply a lack of information about the reaction itself. It should be possible to omit some atom maps or specify partial information for sets of atoms which might end up in a given position in the product. It is never acceptable to force a user to make up data in order to register a reaction. One should only store exactly what is known about the reaction. Atom maps are, by definition ambiguous with respect to the underlying molecules. Atom maps do not appear in the lexical representation of a unique SMILES. They do appear in the lexical representation of an absolute SMILES.
Finally, atom maps are arbitrary class designations; the values of the numbers have no meaning. The Daylight system reserves the right to change the class numbers upon canonicalization of a reaction. The system will reorder the atom map classes over the entire reaction during canonicalization. The resulting maps are guaranteed to have the same meaning as the reaction before canonicalization. Practically, the maps are renumbered as small, dense integers in canonical atom order, but this is not guaranteed. Also, during canonicalization, the atom map classes for agent atoms are removed.
3.5.2 Hydrogens
Hydrogens in reactions are handled as with molecules; they are suppressed unless "special". Recall that for molecules, hydrogens are special if they are: charged, isotopic, bonded to another hydrogen, or multiply bonded. With reactions, there is an additional case which will make a hydrogen special. It is often desirable (eg. 1,5-hydride shift) to store information about the location of hydrogens as part of the atom map of a reaction. Hydrogens with a supplied atom map are considered "special" and these hydrogens are not suppressed. These mapped hydrogens appear explicitly in Absolute SMILES for reactions. Otherwise, atom-mapped hydrogens do not appear in Unique SMILES.
3.6 Acknowledgments
Development of SMILES was initiated by the author, David Weininger, at the Environmental Research Laboratory, U.S.E.P.A., Duluth, MN; the design was completed at Pomona College in Claremont, CA. It was embodied in the Daylight Toolkit with the assistance of Cedar River Software.
Reference
- https://www.daylight.com/dayhtml/doc/theory/theory.smiles.html