想到一个单词,karyotype: 核型; 染色体组型.
基因组浏览器(ucsc genome browser, igv这类)可以交互式地展示基因组数据的可视化结果,染色体的区段信息等等。
有时我们也需要根据自己的数据画一个这类的图,该怎么办呢?这两天就遇到这个问题了。组里面的一位前辈让我根据一些CNV的位置信息,画出它们在染色体上面的示意图。如下:

起初想了下circos图似乎也能表示,比如某个区域一个样本缺失记为1,两个样本缺失记为2......最后画柱状图的圈。但想想又不太合适,因为没人会在circos图里面只画一两个圈,太少了,而且一个圈画24条染色体太紧凑,CNV的位置看不清楚。所以最后还是决定染色体全都铺开展示。找了很久才找到这个R包——karyoploteR,还比较新,是17年发的文章,相关的教程也很少(只找到一两篇)。今天只用到了这个包里面的几个小功能,下面开始介绍。
1. 安装
这是我第一次用Bioconductor上的R包,所以先简单看了一下安装Bioconductor上R包的方法。我知道的是这么几种:install.packages()、source()+biocLite()、BiocManager::install()。
R语言 Bioconductor包的安装指南
不用biocLite安装Bioconductor包——Y叔叔 biobabble
再结合官网说明,安装问题就解决了: http://bioconductor.org/packages/release/bioc/html/karyoploteR.html
(library(karyoploteR)的时候,可能会显示没有某些包,补上就好了)
2. 使用
快速入门: http://bioconductor.org/packages/release/bioc/vignettes/karyoploteR/inst/doc/karyoploteR.html
详细手册: http://bioconductor.org/packages/release/bioc/manuals/karyoploteR/man/karyoploteR.pdf
thus can be used to plot genomic data coming from anywhere. The only exception to this are the ideograms cytobands, that by default are plotted using predownloaded data from UCSC.
可用于绘制来自任何地方的基因组数据。唯一的例外是染色体G显带信息,默认情况下使用来自UCSC预先下载的数据绘制。
2.1 kpPlotRegions
这个绘图函数刚好可以用来画我需要的图,即CNV的位置信息。
举个例子:

gains <- toGRanges(data.frame(chr=c("chr17"), start=c(4000000),end=c(80000000)))
losses <- toGRanges(data.frame(chr=c("chr3"), start=c(80000000),end=c(170000000)))
kp <- plotKaryotype(genome="hg19")
kpPlotRegions(kp, gains, col="#FFAACC")
kpPlotRegions(kp, losses, col="#CCFFAA")
data.frame(): 创建一个数据框;
toGRanges(): 将接收到的表示染色体位置的“表格”转换为R能识别的格式——GRanges;
用法: toGRanges(A, ..., genome=NULL), A可以是(一个或几个)数据框, BED格式的文件, GRanges对象;
GRanges: 存放基因组位置和相关注释的向量。 向量中的每个元素由序列名称,区间,链和可选数据列(例如score,GC含量等)组成。
plotKaryotype(): 创建一个只含有染色体核型和染色体名称的图(最原始的图);
用法:
- plotKaryotype(genome="hg19", plot.type=1, chromosomes=c("chr1","chr2"));
- genome可以是UCSC中认可的基因组名称,比如hg19, mm10等等,也可以是GRanges对象;
- plot.type有1...7可以选,默认是1,具体含义如下:
- plot.type=1 所有染色体依次水平铺开,每个染色体上面有一个画图面板;
- plot.type=2 所有染色体依次水平铺开,每个染色体上下各有一个画图面板;
- plot.type=3 所有染色体在一条水平线上铺开,每个染色体上下各有一个画图面板;
......
其它的几个也好理解,但只能水平画染色体算不算一个小缺点呢?
- chromosomes=c()选择需要展示的染色体,默认情况下全都展示;
- zoom = A可以用来定义显示区间,A可以是数据框, BED格式的文件, GRanges对象,但只有第一个区间会被使用;
- cytobands=A可以用来画自定义的染色体条带(涉及到细胞遗传学的概念),A可以是GRanges对象,数据框, BED。不过表示条带的这一列必须以 "gieStain"命名,值为gneg ,gpos25 ,gpos50,gpos75,gpos100,acen(#red, centromere),stalk(#indented region, region with repeats),gvar等。在查找这一部分内容时,意外发现MATLAB可以画染色体色带图,学过数学建模的小伙伴可以了解一下:https://www.mathworks.com/help/bioinfo/ref/chromosomeplot.html
- main=""可以定义一个图片名称;
以上就是plotKaryotype()的用法。再来看看kpPlotRegions()。
kpPlotRegions(): 根据GRanges对象提供的位置信息,在染色体上画矩形。
用法:
- kpPlotRegions(karyoplot, data, data.panel=1, r0=NULL, r1=NULL, col="black", border=NULL, avoid.overlapping=TRUE, num.layers=NULL, layer.margin=0.05);
- karyoplot: plotKaryotype()的返回值,也就是只含有染色体核型和染色体名称的图;
- data=A表示区间,GRanges对象,数据框, BED都行;
- data.panel=1表示在染色体的上面板画,2表示在染色体的下面板画,具体取值要结合plotKaryotype()函数中的plot.type参数来选择;
- r0, r1表示针对这一行画图命令,利用面板(范围是0-1)的哪一部分,比如r0=0, r1=0.5就是只利用一半面板来画图;
- col, border表示填充颜色和边界颜色;
- avoid.overlapping为真,若区间有重叠则堆叠起来,为假若区间有重叠则取并集只显示一个矩形,默认为真;
- num.layers表示将一个大面板(panel)划分为几个小面板(layers)来画矩形,默认为NULL,会根据最大重叠次数来自动划分panel;
- layer.margin小面板之间的间隔;
2.2 实战
这一个绘图函数应该讲得比较清楚了,下面来应用一下。
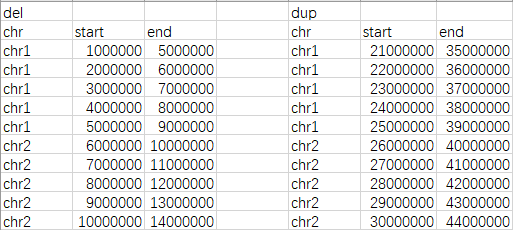
dels <- read.table("del.txt",header = T)
dups <- read.table("dup.txt",header = T)
gains <- toGRanges(dups)
losses <- toGRanges(dels)
my_kp <- plotKaryotype(genome="hg19",plot.type = 2, chromosomes=c("chr1","chr2"),main="my test")
kpPlotRegions(my_kp, gains, col="blue",border = "white",r0 = 0, r1 = 1,num.layers = 5)
kpPlotRegions(my_kp, losses, col="red",border = "white",data.panel = 2)
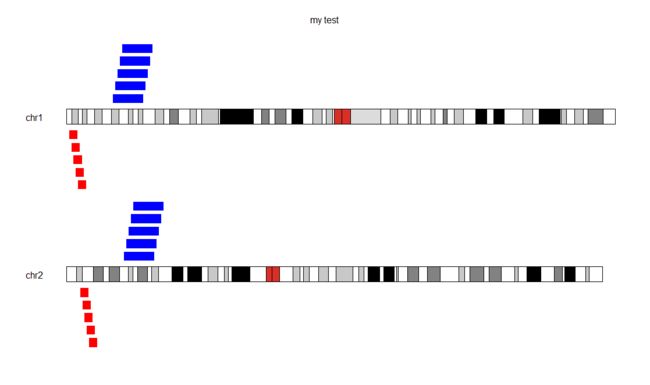
到这儿这一部分就结束了,这个R包的其他绘图函数下次再讲。
另外,看完这些可能会有一个疑问,其他的一些物种可以画吗?按照官方的说明,在UCSC上能找到的应该都能画。不过,一些植物物种应该就只能根据自己的数据绘制核型图了。