从病毒到免疫, “科学地”揭开新冠病毒的神秘面纱

鼠年春节期间,新型冠状病毒(2019-nCoV,后正式命名为 SARS-CoV-2)肆虐,疫情牵动人心。面对不断变化的疫情发展,微软亚洲研究院机器学习组的研究员们基于自己在计算生物学和机器学习等领域的专业知识和研究经验,在第一时间梳理分析了疫情爆发以来科研界针对新冠病毒的研究成果,并尝试利用 AI 技术对新冠病毒进行了初步性的研究探索。
在本文中,研究员们对冠状病毒的基因组与蛋白质结构、免疫应答与分子机理、疫苗与药物研发等进行了介绍,希望能逐步揭开新冠病毒的“神秘面纱”,帮助读者全面了解新冠病毒的致病机理、研发特效药物和疫苗背后的科学问题。同时,研究员们也希望 AI 等计算方法可以与新冠病毒的研究有更深入的结合,为后续的病毒研究带来更多帮助。
一、病毒篇
初识新冠病毒
不同于绝大多数由细胞组成的生物,病毒是一类无细胞结构的简单生物。病毒由负责繁殖后代的遗传物质(核酸)和保护遗传物质的蛋白质外壳组成。不同的病毒通常具有不同形状的蛋白质外壳,而冠状病毒正是因为其外壳像一顶皇冠而得名。2003 年爆发的 SARS(严重急性呼吸道综合征)和 2012 年爆发的 MERS(中东呼吸综合征)都是由冠状病毒引起的。在冠状病毒的结构中,像皇冠一样的刺突称为刺突糖蛋白(Spike Glycoprotein,以下简称 S 蛋白),是结合人体细胞上相应受体的罪魁祸首;E 蛋白是包膜蛋白,将病毒内部的遗传物质包裹起来;还有膜蛋白(M 蛋白)和核衣壳蛋白(N 蛋白)等结构。
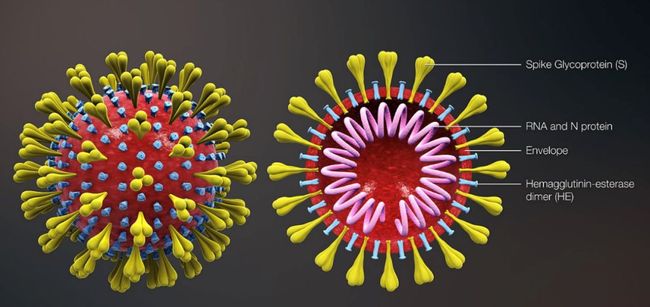
图1:冠状病毒结构示意图
基于序列比对的基因组分析
与我们人类的遗传物质双链 DNA 不同,冠状病毒是一类单链 RNA 病毒。比起稳定的双链 DNA 结构,单链的遗传物质使得冠状病毒具有更强的变异性。冠状病毒家族已知的至少有几十个成员,其中的大多数与我们人类相安无事,而经过多次变异产生的 SARS-CoV,MERS-CoV 等 7 种冠状病毒可以感染人类。近期的研究表明,与 SARS-CoV 和 MERS-CoV 病毒类似,SARS-CoV-2(2019-nCoV)的基因组也分为非结构基因和结构基因两个部分。其中约占 SARS-CoV-2 全基因组总长度三分之二的非结构基因含有两段编码蛋白质的序列,称为 ORF1a 和 ORF1b。而紧随其后的结构基因区域则编码 S 蛋白、E 蛋白等结构蛋白。
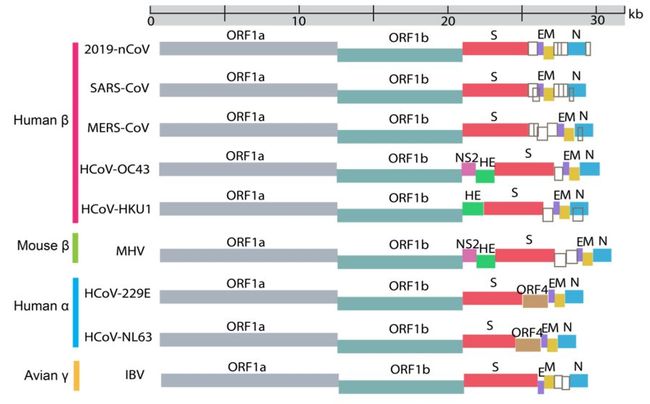
图2:几种冠状病毒基因组的序列比较
序列比对技术是生物信息学对人类社会最为重要的贡献之一。无论是亲缘鉴定、新物种识别还是潜在遗传病分析,都离不开序列比对。序列比对分为局部序列比对和全局序列比对,具有不同的匹配算法。从最简单的动态规划算法基础上,局部序列比对开发出“水人算法”(Smith–Waterman algorithm),全局序列比对则采用“针人算法”(Needleman–Wunsch algorithm)。从全基因组结构上分析,SARS-CoV-2 (2019-nCoV) 的基因组结构与其它冠状病毒比较相似。它与发现自蝙蝠的两种冠状病毒(Bat-SL-CoVZC45 和 Bat-SL-CoVZXC21)的相似度分别为 87.5% 和 87.6%;与 SARS 病毒的序列相似度约为 79%;而与 MERS 病毒的序列相似度仅有 50%。值得注意的是,SARS-CoV-2 与 SARS-CoV 在基因组的不同区域的序列相似性不尽相同:在编码病毒与宿主受体结合的 S 蛋白区域,SARS-CoV-2 与 SARS-CoV 的序列相似性显著降低。
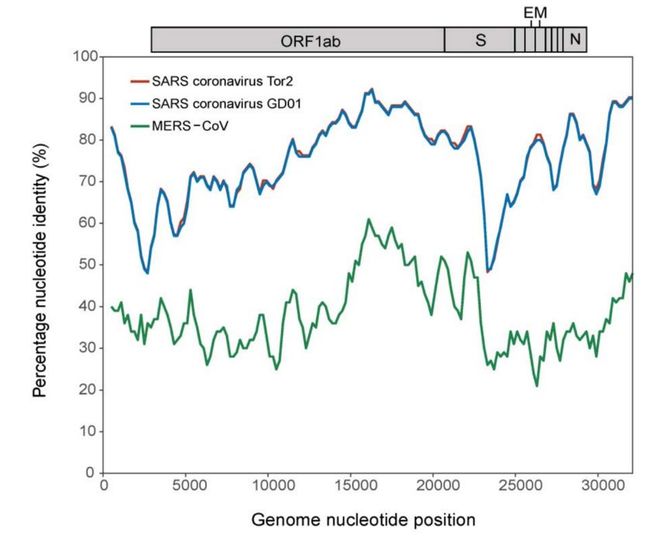
图3:SARS-CoV-2 与 SARS-CoV、MERS-CoV 基因组序列相似性的比较
冠状病毒的进化
基于层次聚类的进化关系分析
基于序列比对的结果,我们还可以进行层次化聚类,从而得到不同病毒之间的亲疏关系和进化路径。依照下图所示的冠状病毒序列进化树,我们可以看出 SARS-CoV-2 与另外两种冠状病毒 Bat-SL-CoVZC45 以及 Bat-SL-CoVZXC21 在全基因组的进化关系分析中最为亲近。进一步分析,在 7 种已知可致病的冠状病毒中,SARS-CoV 与 SARS-CoV-2 的进化关系最近,而与造成较高致死率的中东呼吸综合征病毒 MERS-CoV 的进化关系较远[4]。这一研究表明 SARS-CoV-2 与 SARS-CoV 具有较为保守的进化关系,从而暗示新型冠状病毒在受体结合以及致病机理等方面可能与 SARS-CoV 有诸多共通之处。
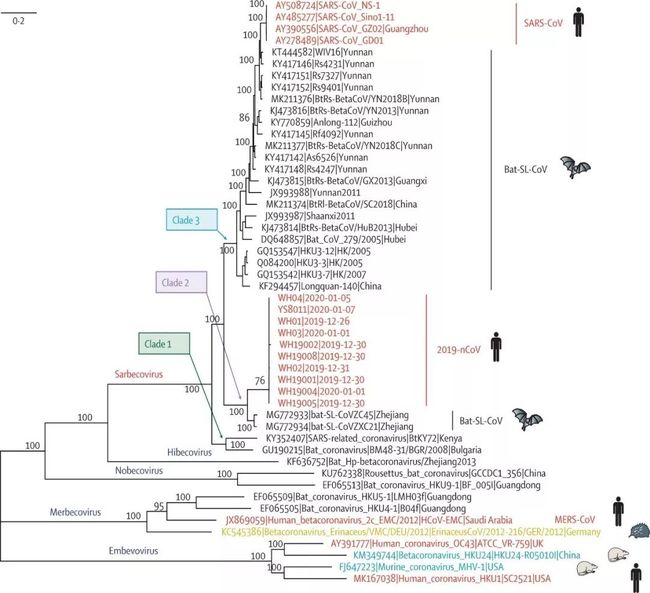
图4:SARS-CoV-2(2019-nCoV)进化分析(左上为进化标尺)
新冠病毒结构的解析与预测
基于深度学习的蛋白质结构预测
如果把基因组序列比喻为标识一个人的身份信息,蛋白质的三维结构就是一个人的身形容貌。三维结构的解析对于新冠病毒致病机理和药物设计具有非常重要的先决意义。令人欣喜的是我国科学家已经解析出 SARS-CoV-2 非结构区域基因对应的高分辨率三维蛋白结构,而对于编码结构蛋白的基因区域,我们在拿到新冠病毒的基因序列后,第一时间利用我们基于深度学习技术自主开发的结构预测软件进行了三维结构的预测。对于 SARS-CoV-2,S 蛋白区域无疑是我们最为关注的一个部分。针对于 S 蛋白和某个已知结构的模板蛋白,我们首先使用深度学习技术预测单个氨基酸残基和成对氨基酸残基的基本特征,然后根据预测的特征计算两个蛋白比对打分,最后通过交替方向乘子法求解出两者之间最优比对。我们通过搜索结构库中所有蛋白,选择出最佳结构模板以及计算出两者最优比对,进而以此为基础利用常用的结构建模软件预测出 S 蛋白的三维结构。
我们将预测的 S 蛋白结构(下图左边蓝色结构)与 SARS-CoV 的 S 蛋白(下图左边红色结构)进行了结构联配,结果表明预测的 S 蛋白与 SARS-CoV 对应区域的结构高度相似。根据以上结果,我们推测虽然 SARS-CoV-2 与 SARS-CoV 在 S 蛋白区域的基因序列相似性不高,但二者在蛋白质的真实结构上可能比较类似。此外,我们预测的 S 蛋白结构(下图右边蓝色结构)与密歇根大学 Yang Zhang 组利用 C-I-TASSER 软件预测的三维结构[5](下图右边红色结构)做了比较,除了左侧几个螺旋结构有所不同,两个研究组预测的整体结构非常相似。
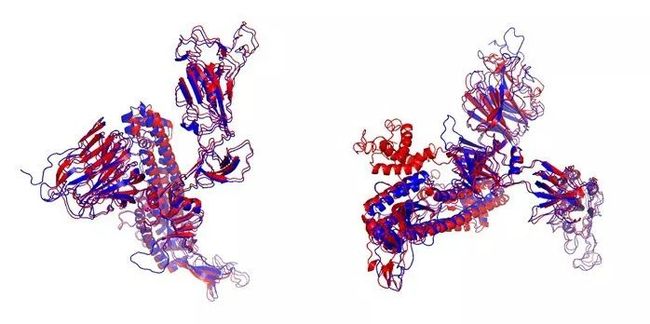
图5:SARS-CoV-2 的 S 蛋白预测结构与 SARS-CoV 中对应结构(左, PDB ID: 5X58A)以及 C-I-TASSER 预测结果[5] (右) 的比较
新冠病毒与宿主结合机制推断
基于分子动力学模拟的受体结合分析
冠状病毒与宿主细胞受体结合的位点通常位于 S 蛋白上的部分区域(称为亚结构域)。不同种类的病毒其对应结合的受体也不尽相同。例如,SARS-CoV 的结合受体为 ACE2 (血管紧张素转化酶2)。如前文所述,根据我们的三维结构预测结果,新型冠状病毒的 S 蛋白结构可能与 SARS-CoV 的较为类似。对 S 蛋白预测的三维结构进一步分析表明虽然部分氨基酸残基与 SARS 病毒中对应的氨基酸残基有所不同,但是 SARS-CoV-2 的 S 蛋白中潜在的负责与受体结合的亚结构域与 SARS-CoV 蛋白中对应的亚结构域非常类似。最新研究发现,SARS-CoV-2 的 S 蛋白和 ACE2 具有较高的结合强度,表明 ACE2 非常可能是 SARS-CoV-2 的宿主细胞受体[3][6]。在后续的研究中,我们期待有更多的利用分子动力学模拟、分子对接等计算生物学技术手段对 SARS-CoV-2 与其对应的宿主受体的结合过程、构象变化、亲和力和自由能等方面展开更为深入的研究,以及对 SARS-CoV、MERS-CoV 等重要冠状病毒的致病机制在分子层面更为细致的阐述与比较。这些研究将加深我们对冠状病毒的认识,对设计药物和疫苗具有重要的价值和意义。
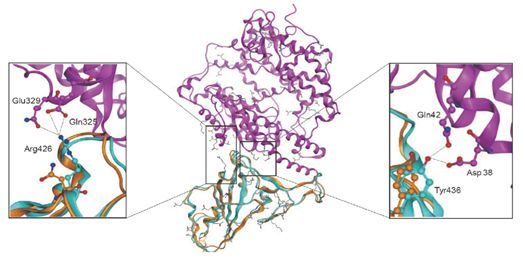
SARS-Cov-2 的 S 蛋白同源建模结构与 ACE2 互作示意图
二、免疫篇
人体免疫系统是由多个器官、多种免疫细胞以及各种免疫分子构成的一个复杂系统。它们通力合作,构建起防御各种病原体(病毒、细菌、寄生虫等)的层层防线。人体免疫系统分为固有免疫和适应性免疫系统。其中,固有免疫也被称为非特异性免疫,可以对入侵的病原体快速作出反应。比如固有免疫系统中的巨噬细胞、中粒性细胞通常可以在第一时间达到人体发生炎症的地方,吞噬病原体或清理死亡的人体细胞。此外,固有免疫系统还会通过抗原呈递等方式激活人体的特异性免疫应答。
适应性免疫系统
固有免疫是非特异性免疫系统,要想更加高效地对付某种特定病原体则需要通过适应性免疫系统产生特异性免疫应答来发挥作用。大家平时所关心的抗体或者疫苗,都与其有着密切的联系。B 细胞和 T 细胞是适应性免疫中的主要“兵种”。与固有免疫中不同的是,这些兵种的战士可以识别并消灭某一种特定的病原体。同时,有些士兵还能记住目标敌人的样子,一旦相同的敌人再次入侵,它们就可以快速拉响警报,发起对入侵者的剿灭战。
成熟的 B 细胞会携带一种被称作 B 细胞受体的探测器,一旦侦测到相应的抗原,并在辅助 T 细胞的帮助下,它们就会进行增殖分化。一部分分化成能够产生抗体的浆细胞,另一部分变成记忆 B 细胞。抗体具有和产生它的 B 细胞一样的探测器,在体液中巡逻,并标记那些特定的病原体或直接阻碍它们感染人体细胞。疫苗正是利用了这一机制,通过来自病原体的抗原信息激发 B 细胞产生抗体防御外敌。
T 细胞是另一类重要的特异性免疫细胞。辅助 T 细胞的主要功能是在识别抗原之后,通过释放细胞因子来调控或辅助其他免疫细胞发挥作用,比如协助激活 B 细胞、活化杀手 T 细胞等。杀手 T 细胞则会瞄准那些带有特定抗原信息的受感染细胞,通过释放细胞毒素来杀死他们。T 细胞和 B 细胞一样,也是利用一种被称作 T 细胞受体的探测器来识别特定抗原。
杀手 T 细胞在毒杀癌细胞
抗原和抗体
抗原是指那些能够激起机体免疫应答并能被特异性免疫产物识别的物质。当 B 细胞受体或 T 细胞受体能够和抗原上的某些部分结合时,就完成了对这个抗原的识别。这些能够被结合的部分被称为抗原决定簇或抗原表位。
对于一个抗原而言,并不是它身上的任何部位都能够成为探测器的靶子。并且,由于个体之间的基因差异,能够成为靶子的抗原表位在不同个体之间也可以是不同的。正是因为这一点,研发疫苗过程中一个很重要的步骤就是去确定抗原上能够成为靶子的部位,同时尽可能寻找对不同人群都起作用的那些靶子。
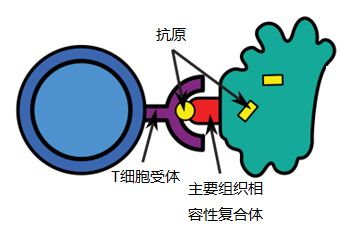
T 细胞受体识别抗原表位示意图
1、基于数据库检索和序列比对的抗原表位预测
那么新冠病毒上的靶子有哪些呢?这些靶子能够覆盖多少人群呢?香港科技大学的 Syed Faraz Ahmed 等研究人员利用已有的关于 SARS 病毒的抗原表位数据,筛选出了 268 个 T 细胞抗原表位候选,这些抗原表位在当前已经公布的新冠病毒蛋白质序列中均可以找到,并估计可以覆盖约 96.29% 的全球人口和 88.11% 的中国人[9]。这些候选抗原表位可以为新冠病毒疫苗的研发提供有用的信息。
T 细胞抗原表位在 SARS-CoV S 蛋白中的位置
2、基于深度学习的抗原-T 细胞受体绑定预测
随着高通量测序和免疫实验技术的发展,越来越多的基因和免疫实验数据可以被生物学家和计算机科学家利用,通过计算方法来理解免疫系统、研发疫苗、以及辅助疾病的诊断和治疗。微软亚洲研究院机器学习组的研究员一直在利用深度学习技术积极开展有关抗原识别的一系列工作。例如,在抗原的多个靶子中,预测哪些能够激发强免疫反应;给定一个抗原靶子的时候,预测哪些 T 细胞能够识别它。生物学家通过高通量的免疫实验筛选出与某种抗原产生免疫反应的 T 细胞,并通过高通量测序技术测定出这些 T 细胞受体的 DNA 序列,这样就产生出了一组抗原-T 细胞受体的绑定数据。随着这些实验数据的不断产生和积累,我们就有机会利用机器学习技术对其进行建模从而帮助解释 T 细胞受体和抗原绑定的规律。这方面的研究工作目前还处于起步阶段,由于数据规模相对有限,模型的泛化能力还有待进一步的提高。
3、基于免疫实验方法的抗体分析
当一个病毒感染者被治愈后,只要其免疫系统中的 B 细胞被激活,体内通常会存在抗体。对于新冠病毒而言,它会在人体中产生什么样的抗体呢?全面回答这个问题还有待进一步的研究。复旦大学医学院的应天雷教授团队在这方面有了初步的成果[10]。新型冠状病毒和 SARS 冠状病毒在 RNA 序列和蛋白结构上有比较高的相似度,并且都可以通过刺突蛋白与 ACE2 受体结合来感染人体细胞。因此,他们分析了已知的几个针对 SARS 冠状病毒刺突蛋白的抗体,通过免疫实验发现其中的 CR3022 很可能也是针对新型冠状病毒刺突蛋白的抗体之一。
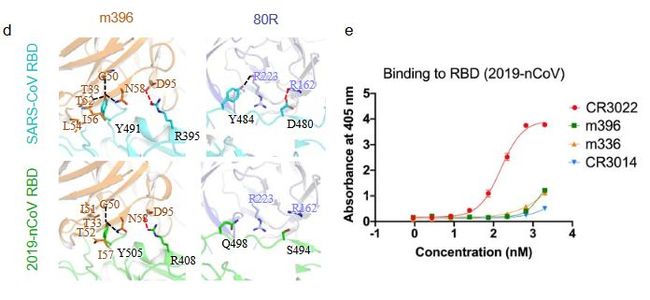
SARS-CoV RBD 及 2019-nCoV RBD 与抗体复合结构(左)及抗体与 2019-nCoV 的 ELISA 实验结果(右)
抗体在目前的疾病诊断与治疗上有着不同程度的应用。利用抗体辅助诊断已经是临床疾病诊断中的常见手段,但是在疾病的治疗上,由于受到纯化、储存、有效性等方面的制约,目前的临床应用还相当有限。值得一提的是,蛋白质结构预测、分子动力学模拟等计算技术正在越来越多的被应用到抗体的发现和设计中。
三、药物和疫苗篇
药物研发
1、干湿实验结合的药物筛选
特效药物研发是“永远在路上”的科研攻关。目前还没有完全解析 SARS-CoV-2 的全部蛋白结构,对其与受体结合分子机理的研究方兴未艾,但我们欣喜地看到很多研究表明多种治疗其它病毒的药物对 SARS-CoV-2 具有潜在的治疗作用。由蒋华良院士、饶子和院士领衔的科研团队,在解析 SARS-CoV-2 的 ORF1ab 区域后,通过计算机模拟和生化实验验证相结合的手段筛选出 30 种可能对 SARS-CoV-2 有治疗作用的药物分子。这些潜在的药物多为蛋白酶抑制剂,对病毒繁殖的抑制、阻断病毒与受体细胞结合方面具有潜在的功效。另据报道,用于抵抗埃博拉病毒的试验药物 Remdesivir(RDV)可能具有抗 SARS-CoV-2 的功效。作为一款新型核苷类似物抗病毒药,多个研究组的报道显示有病人在服用 RDV 后发热、咳嗽等临床症状有所减轻,病情出现好转。
2、基于互作网络分析和分子动力学模拟的高通量药物筛选
除了以上通过生物实验和临床测试等手段筛选药物,结合机器学习和人工智能等技术,利用高通量的计算机辅助药物设计和预测也在“老药新用”的策略中大展拳脚。通过生物医学的实验手段研发药物,主要是对已经批准上市的广谱抗病毒药物进行测试和筛选。这类药物以抗病毒的干扰素和阻碍病毒侵入宿主细胞的信号通路类药物为主,其作用主要是通过抑制病毒的逆转录、复制等过程和增强人体自身的免疫应答实现。与临床实验筛选不同,通过计算手段进行的药物筛选可以在更广的范围上进行高通量筛选。
Docking 模拟是分子模拟的重要方法之一。其本质是两个或多个分子之间的识别过程,是预测蛋白质结构与有机小分子结合的常用计算模拟手段。从经典的牛顿力场,到通过机器学习算法设计高效、简化的量子力场,Docking 模拟的准确性和效率都得到显著的提升。Docking 模拟可以快速分析不同小分子与蛋白质结合的作用位点和结合作用方式,解释药物作用靶点发挥药效的原因,为计算机辅助药物筛选提供指导。通过 SARS-CoV-2 蛋白结构与已知的有机小分子药物库中的海量候选分子进行 Docking 模拟,分析不同分子与病毒蛋白结合的自由能和亲和力,高通量地筛选出抵御病毒的潜在药物分子。目前国内外已经有多个研究组采用这种技术筛选出洛匹那韦(Lopinavir)等抗 HIV 药物可能具有抵御新冠病毒的潜力。此外,利用生物信息学的分析手段,从 DrugBank、Therapeutic Target、PharmGKB 等多个数据库中提取已知的药物-靶点互作信息,构建药物-靶点互作网络,通过已知的作用于冠状病毒靶点的药物预测对 SARS-CoV-2 具有潜在作用的药物分子。有研究表明,通过构建基于系统药理学的网络,量化冠状病毒与药物靶点之间的相互作用,已经找出西罗莫司加放线菌素、巯基嘌呤加褪黑素、托瑞米芬加大黄素等多种潜在的药物组合[11]。虽然计算机辅助的“老药新用”可以高通量地筛选潜在药物,但真实的药效、副作用等关键因素仍然需要严格的临床试验才能得到验证。
“老药新用”是快速筛选药物的手段,而针对 SARS-CoV-2 的特效新药的研发则是对新冠病毒的精准打击。然而,新药的研发通常需要极为漫长的流程。而随着深度学习技术在蛋白质结构预测和蛋白质小肽药物设计等领域的深入应用,新药研发的效率将得到有效提升。我们相信随着“老药新用”在临床上的不断尝试,以及针对 SARS-CoV-2 特效新药的快速研发,缓解和治疗 SARS-CoV-2 的药物会陆续被研发上市。

部分潜在的抵抗 SARS-CoV-2 的药物
疫苗研发
目前还没有针对新冠病毒的疫苗问世。疫苗按照不同的分类方法和制备工艺,可以分为很多类别。但是疫苗的基本原理和作用是一致的,即利用抗原本身来制备生物制剂,通过接种到人体来激活免疫反应,比如生成抗体,从而使得人们在将来遇到真正的相同病原体时,可以及时地发现和消灭它们。
疫苗需要具有几个基本特性。第一是安全。疫苗既然来源于抗原,比如新冠病毒,它必须经过减毒等措施,使得被接种到人体之后不会致病或引起不良反应。第二是有效。疫苗要能够促使 B 细胞产生抗体,并在下一次遇到相同的抗原时,抗体能够准确识别。第三是普适。疫苗是一种公共卫生干预措施,疫苗研制的目标之一就是希望制备出的疫苗能够尽可能多的覆盖人群。
疫苗的研制是一个非常复杂、严格和漫长的过程。以传统病毒疫苗为例,通常需要经过毒株的筛选、减毒、繁殖、过滤、提取等过程;在建立动物模型、完成动物试验并取得申报许可后,才能进入共三期的临床试验阶段;最后还需要通过专家评审和国家批准方可投入生产并最终上市[12]。此外,疫苗是具有特异性的,而病毒是不断进化的。这意味着一种疫苗在病毒发生变异之后可能就会失效。这也是为什么世界卫生组织会积极跟踪各类病毒,尤其是流感病毒的进化,并积极推动降低疫苗研制和生产周期与成本的相关研究。
针对新冠病毒的疫苗研发正在如火如荼地进行着。虽然最新的 mRNA 疫苗技术可以大大缩短疫苗的研发周期,但是为了严格保证安全和有效性,动物试验、临床试验等步骤仍然不可或缺。
四、展望
当下,对病毒的抗击仍未结束。病毒藏其形于微小,人类必治之以群智。面对这场病毒对人类的“突袭”,各行业倾力合作,共同抗击。医疗和生物研究中产生的海量数据蕴藏了非常有价值的信息和知识,而 AI 技术在知识提取和建模分析中表现出了巨大潜力。我们在本文中展示了 AI 技术在序列分析、结构预测、免疫学研究和药物研发等方面的诸多应用。展望未来,医学、生物、计算机等研究机构在数据和技术上的共享与合作将更加深入,继续为人类健康保驾护航。风雨同舟,爱相随,AI 相伴!
本文作者:王童*、夏欢欢*、朱建伟、何亮、邵斌、刘铁岩
(*同等贡献)