本文翻译自拜耳医药全球药物研发靶点研发的What makes a good drug target,文章不长,提供了药企视角下的药物靶点。
医疗缺口很大的领域需要创新的治疗方法,而这基本依赖于新的药物靶点的发现。尽管生物药大大拓展了可以成药的分子范围,合适的药物靶点依然缺乏。发现并评估药物靶点的医用价值,不仅需要大量的实验性、机制和药理学研究,还需要理论分子成药性分析、可能副作用的早期分析、市场需求分析等等。本文旨在定义一个医药公司视角下的好的药物靶点的性质。
前言
近来对中止研发的药物产品和临床失败案例的分析发现,越来越多的化合物因无法通过药效终点而夭折。新项目通过二期临床的成功率从2006-2007年的28%下降到了2008-2009年的18%。汤森路透生命科学咨询公司分析了2008年至2010年108例有报道的临床二期失败案例,包括新药以及已经上市药物的新适应症,发现有51%药物失败的原因是药效不足。而且,对2007年至2010年间III期临床以及申报失败的分析也发现医疗领域中三分之二的失败原因为药效不足。因此,经体内、体外模型预测,新的、有前景的、具备临床药效可能性的药物靶点是药物发现成功的关键。
2009年,拜耳医药提出了“Grant4Targets”倡议。其基本观点是为学界提供桥梁和药物发现技术来支持新的药物靶点的评估和验证。尽管我们收到了很多高质量的、有价值新药靶点的提议,却也越来越明白对于“什么是一个好的新药靶点的关键性质”我们还没有讨论清楚、没有清晰定义。
所以,本文旨在从医药公司的视角定义创新的、有前景的药物靶点所需的基本条件。我们将重点讲述药物靶点的确立、挑选、评价、验证过程中的重要方面。另外,我们还特别强调在该过程的早期就要重点考虑成药性和可验证性。此外,我们还对未来可能出现的提升药物成药性空间的技术做了展望。最后,我们希望新药物靶点的定义能够很大程度上依赖于有着丰富医疗需求和不同药物安全性要求的医疗领域和特定适应症。
理想药物靶点的性质:
具备疾病修饰和/或有证明有效的病理学证据
靶点的修饰对正常生理或其他病症下的生理环境影响不大
如果成药性不明确(如激酶靶点的药物),要能够获得靶蛋白的3D结构或者类似结构进行成药性分析
具有好的可验证性,能够做高通量筛选
在人体不同组织内呈现差异化表达
存在可以用来检测药效的靶点/疾病特异性的biomarker
通过表型数据(如基因敲除小鼠或基因突变数据库)较容易推测潜在的副作用
靶点具备较好的IP状况(没有竞争性靶点,没有限制)
靶点定义和分类
尽管跨越多条通路的关键分子在靶点发现中很重要,也取得了很大的成功,但是,对多个通路分子靶点都有效的化合物(hit)往往也有严重的副作用。所以,对靶点的高度特异性识别左右着药物研发的进程。
一个药物靶点通常是蛋白、多肽或者核酸,该靶点的活性可以被药物——小分子SMOL,或抗体/重组蛋白等大分子BIOL——修饰而发生改变。2006年针对所有申报成功的医疗药物,科学家对324个药物靶点的总数达成共识。采用生物信息学的手段,Bakheet和Doig从3573个非靶点分子中筛选出了668个具备靶点性质、可能成为新靶点的蛋白。虽然目前绝大多数的药物发现中的靶点都是蛋白,核酸靶点将会越来越重要。
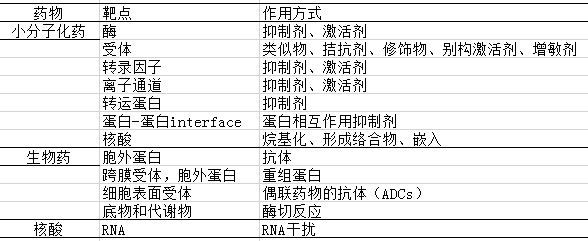
小分子药物靶点
SMOL药物靶点主要为蛋白类:酶、胞外/核受体、离子通道、转运体等。目前已经应用DrugBank数据库对药物靶点家族的增长进行了分析。该数据库被认为是药物和药物靶点信息最重要的来源之一。Rask Andersen等人分析了2242组药物-靶点相互作用,发现了人基因组中有435个效应介导的药物靶点,可以被989个不同的药物所修饰。所有药物靶点中,193个为受体蛋白,占比44%。其中GPCR是降压药物和抗过敏药物常用靶点,占所有药物靶点的比例为36%。
除去已经建立的药物靶点类型,新的技术手段也应用到原本无法成药的靶点上,如蛋白-蛋白相互作用。Moellering等人研究表明直接与SAHM1的结合可以阻止活化的NOTCH-癌蛋白复合物的形成。近来有报道成功地使用竞争性抑制剂的方法,抑制了组蛋白和BRD蛋白的相互作用,把BRD4融合的癌蛋白从染色质上解离下来,进而达到控制肿瘤生长或调解炎症进程的目的。也有前沿化学技术开发出小分子化合物活化酶活性的新机制。未来,新技术手段还会不断拓展成药性的空间,新的小分子药物靶点还会发展出来。
生物药靶点
由于抗体或重组蛋白类药物无法进入细胞,胞外蛋白和细胞表面受体是大分子药物的合适靶点。大多数开发的抗体药物都是针对癌症和炎症类疾病的。ADCs属于新生物药发展的互补策略。如基因泰克的曲妥珠单抗-美坦辛(T-DM1),就结合了单抗药物和微管抑制剂的优势,目前已经进入多临床实验。ADC策略中,靶点不需要具备修饰影响细胞的功能,因为细胞毒试剂会杀伤细胞,而抗体把细胞毒试剂定位到表达抗原的病理组织。
靶点评估
下图详细描述了拜尔医疗的靶点评估过程。新药物靶点成功确立之后,需要进行详细的分子靶点评估:根据疾病假设设计实验研究药代动力学性质、分子成药性的理论分析以及潜在靶点相关biomarkers的预判。
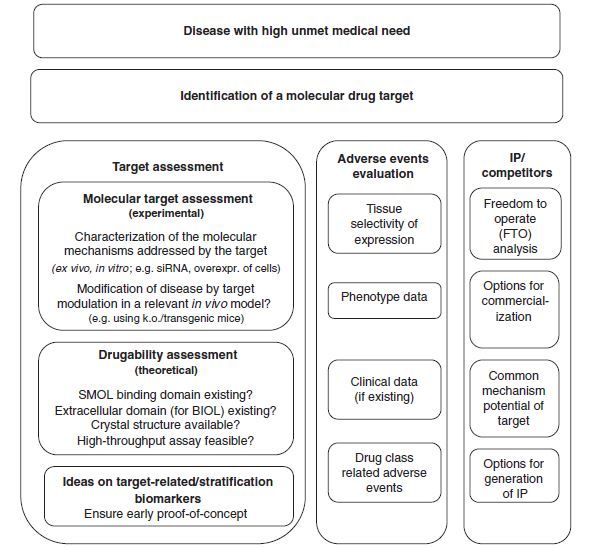
靶点确立
靶点评估的第一步。新靶点的最大来源是相关文献,因为全世界无数的科学家都在寻找新的分子通路以及基因和蛋白的新功能。文献之外,最有价值的参考是分析目标组织或者比较正常组织与疾病组织中RNA和/或蛋白的表达。当综合各信号通路分析、强大的整合数据,这些表达数据能够提供比RNA或蛋白正常调控水平下更多的潜在靶点的信息。就蛋白质组学而言,基于活性的蛋白图谱(ABPP)可以比较出疾病和正常组织中酶活力水平的不同,从而确立靶点,进而大大拓宽了靶点确立范围。还有一种新药物靶点信息的来源基于敲除小鼠基因改变和表型变化、体细胞突变、基因融合以及拷贝数变化等等,这些信息来源更适用于癌症靶点。

如果针对靶点的药物是抑制剂或激活剂,功能基因组学结合表型筛选更有优势,因为在细胞模型体系中建立靶点,再进行挑选/改造表现出疾病模型的过程更便捷。细胞模型中,干扰基因表达(如siRNA或shRNA,过表达cDNA,或者使用小分子抑制基因功能)和合适的表型读取系统联合使用非常强大。众多例子中,高通量RNA干扰筛选技术已经成功地应用于靶点确认。化合物基因组学方法通常不够直接,因为表型筛选之后还需要靶点解码。参考文献[21,22,24,-28]对支持药物靶点确认的复杂的数据库有更为详细的描述。在拜耳医疗,我们把公开的以及Phylosopher数据库关于靶点的全部信息整合在一起,所有拜耳的科学家可以共享。靶点确认时,与整合后数据互补的信息被优先参考。不过,当采用公开数据时,关键结果会在内部被重复,因为我们发现仅有30%报道的体内和体外实验结果可被重复。
下图列出了成功确立靶点的三个关键要素:对疾病的深入理解、关于分子机制的知识以及预测模型和技术上的支持。对疾病了解得越多,预测模型的支持性越好。我们认为,深入理解病理学和引发疾病的分子机制,应用相关靶点确立和验证技术是有前景药物靶点的确立的基础。
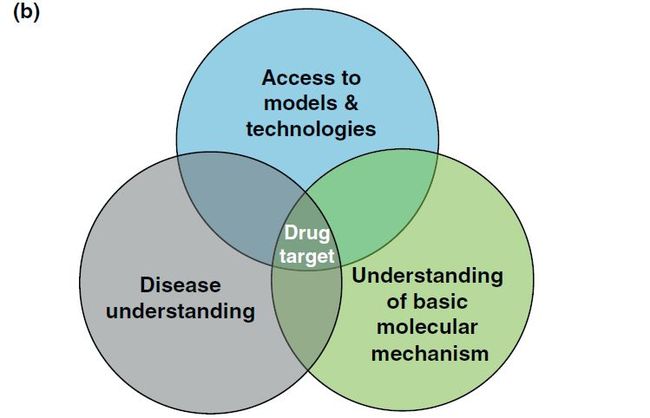
靶点验证
药物靶点必须按照既定的作用方式通过实验加以验证。这里的数据与临床起效的可能性直接相关(如重要人源细胞和组织实验)。这类功能性实验包括基因下调、基因敲除,如果有SMOL化合物库或者工具抗体的话,还可以使用这些特定的工具。体外细胞机制研究可被用来揭示靶点的调控性质以及其所在的信号通路。最后,依据疾病种类的不同,还可以使用合适的动物模型来评估靶点和疾病的相关性。如果小鼠具有与人类同源的功能基因,而合适的疾病模型也存在的话,也可以使用基因敲除小鼠或者转基因小鼠进行靶点验证。然而,总的来说,经过验证的靶点在进行人体验证时也不是没有风险的。因为一些模型对于人体内情况有高度预测性,而另外一些则有很大的偏差。而且,有些疾病是高级灵长类动物固有的,而绝大多数的动物实验都使用老鼠做实验。因此,不是所有风险在早期药物发现过程中都能够被预测。
把上述考虑应用到真正的药物发现实际中来,IL-2诱导的酪氨酸激酶(Itk)可以说是炎症性皮肤病治疗领域的新靶点。Itk在病症组织中特异性表达,主要存在于T细胞,且在皮炎病人受损皮肤中表达升高。采用RNA沉默策略可进行靶点验证,体内实验(Itk敲除小鼠模型)同样可行。另一方面,证实该激酶在疾病模型中作用的SMOL抑制剂也已经得到确认。
成药性评估
目前蛋白靶点的成药性评估方法一般依据蛋白序列相关性质和蛋白的3D结构,文献[24][33]中有描述。已知3D结构的药物靶点可以在潜在药物靶点数据库中找到,目前该数据库涵盖了1207条目、841中已知/潜在药物靶点,结构信息来自PDB(Protein Data Bank)。对于药物靶点评估来说,知道3D结构具有巨大优势,因为可以使用结构成药性搜索引擎(EMBL-EBI)来预测是否具有SMOLs结合位点。
对SMOL结合靶点来说,除了成药性预测之外,还要分析靶点的催化和/或功能因素,还要分析靶点的组织选择性以及具有类似结合位点的其他蛋白。考虑到蛋白的3D结构,各种算法被开发出来用来评估配体设计所需的蛋白结合位点(参考文献[35])。
可验证性评估
为后续筛选先导化合物需要,建立能够确认靶点结合能力和功能的生化和/或细胞学实验十分必要,该过程我们称之为药物靶点的“assayablility”。是否能建立起有意义的方法,取决于靶点类型和关于靶点的信息。因为很多GPCRs的活性可以通过检测第二信使水平的方式来监测,所以一个能够检测下游产物的GPCR的可验证预期也不错,而无法检测下游产物的GPCR,尽管成药性毋庸置疑,但是可验证性就比较低了。在这种情况下,就需要提供合乎药理学相关实验规范的功能性研究方案了。
适应症评估
考察药物靶点还有一个重要的方面就是起适用的临床症状。拜耳有一个抗肿瘤药物——激酶抑制剂sorafenib索拉菲尼(Nexavar/多吉美),其成功至少部分得益于它可以抑制多个激酶活性(包括丝氨酸/苏氨酸激酶Raf和ERK通路)的性质。虽然已有文献数据证实Raf/MEK/ERK1/2活性与心肌肥大的相关性,而心肌肥大是心律失常、心脏病和猝死等病症的危险信号。不过,心肌肥大是心脏病(心肌梗死或持续的血压升高)发展过程中早期的适应不良症状。使用激酶抑制剂预防心肌肥大需要在治疗早期即开始,并很有可能持续一生以防止疾病进展。
很显然,从安全性和顺应性角度考虑,用激酶抑制剂预防疾病所面临的挑战远比用来治疗威胁生命的癌症要大得多。出于同样的考虑,PubMed使用关键字“激酶&抑制剂&心血管&毒性”进行文章搜索,找到了200篇文献(截止2011年6月)提及激酶抑制剂作为抗癌药物在临床上最大的副作用即心脏毒性。因此,有7个FDA批准的激酶抑制剂收到了FDA考虑可能的心脏毒性的警告。信号通路中多个激酶活性可能会导致心肌肥大,但它们也是维持正常细胞功能的重要组分,而另一方面抗癌的小分子激酶抑制剂的靶点特异性不强,这两点或许可以解释该类型药物的副作用。因为激酶中的ATP结合位点非常保守,即使使用复杂的结构生物学工具也很难开发出高特异性的激酶抑制剂。如激酶抑制剂表现出来的SMOL药物的靶点特异性不高的问题,提示我们,BIOL药物——无论是单抗、重组蛋白或者反义核酸和RNA干扰都有更好的特异性——会是更有吸引力的选择。不过,BIOL药物都有无法口服的缺点,在某些适应症中会是大问题。最后,BIOL药物的成本比SMOL高很多,在需要长时间治疗才能获得临床受益的情况下,成本也会是一个难以逾越的障碍。
综上,更多的医疗情况需要特异性的治疗方案,而且,基于目前对疾病的理解,是采用多靶点策略还是选择一个药物一个靶点的方案,还需要仔细评估。
临床和商业需求
临床的高度需求(无药可用或者现有治疗手段在疗效和/或安全性上极大受限)和可预期的市场是还需要考虑的重要因素。
一个药物靶点的医疗潜力在它被发现的初期通常并不显现。一个有名的例子是胆固醇合成通路的限速酶,HMG-CoA还原酶,该靶点在医药产业的销售额是目前已知靶点销售额之冠,但在研发初期,该靶点的第一个抑制剂“compactin”却没有起到降低大鼠胆固醇的作用,因为它引发了大鼠肝脏大量代偿性生成HMG-CoA还原酶,很多研究者因此误入歧途。从抑制HMG-CoA还原酶降低胆固醇的概念提出,到最终为临床所证实,研究者花费了20年的时间。有时候,靶点修饰过程中“失败越早花钱越少”,在靶点确认早期阶段,最好就通过表型学体外实验或最晚在体内实验时就看到药物的疗效。
即使一个靶点已经确认了适应症,研究其在其他适应症领域中的作用并扩大其医疗应用还是很有必要的,也是第一张图中common mechanism potential所指的内容。如果药物在开发晚期遇到巨大困难,还可以选择其他的适应症进行研发。
知识产权
如文献[43/44]所述,药企对价值链的贡献在于最终成药的小分子化合物(或BIOL)的专利而非靶点本身。理想但通常做不到的是两者都保护起来:专利保护的化合物(BIOL或SMOL)和专利保护的对治疗特定疾病的靶点修饰物的使用。
早期的FTO分析保证了特定的研究,如靶点修饰物的研发确认等,在不侵犯他人知识产品的情况下得以进行。
新的靶点提供了这样的机会——如果化学空间允许——为可以和靶点结合的SMOL化合物申请专利,从而获得知识产权保护。而只有当公司获得靶点修饰物的特许经营权,并收取到他们为市售药物付出努力的专利使用费,投资才算获得完全的回报。对于化合物和/或抗体药物都是类似的情况:如果我们没有专利保护的SMOL或BIOL,我们就没有办法应对竞争者,保护自身的利益。
一般来说,因为很多靶点最初都是研究文献报道出来的,对一个给定的靶点来说,它的确认程度和竞争度是直接相关的。一些公司不喜欢在选择靶点时冒险而更乐于接受较大的竞争,其他一些公司却愿意冒险选择那些稍有竞争的靶点,以期研发出一类药。
潜在副作用的早期评估
由于靶点的多效性,同一靶点在不同组织中功能可能不同,或在不同的生长发育阶段表现出不同的功能。所以,检测靶点在人体组织中的表达水平会很有帮助。尽管有例外,一般认为靶点表达范围越广,针对该靶点的药物的副作用就越大,因为药物是作用于整个人体系统的。评估潜在靶点相关副作用的另外一个参考信息是,靶点在疾病组织和正常组织之间的表达差异。举例来说,用于治疗胃返流疾病的质子泵抑制剂就有很高的剂量接受度,因为其分子靶标胃H+/K+ ATP酶几乎只存在于胃粘膜组织。这些描述表症的重要性因个体指症不同而不同。很显然,威胁生命的癌症疾病对靶点相关副作用的忍受度远远高于不那么危险的疾病。
靶点敲除小鼠和人体靶点基因缺陷可以为靶点相关副作用提供更多提示。例如,用来治疗类风湿性关节炎的线粒体酶二氢乳清酸脱氢酶(DHODH)在小鼠实验中发现了致畸性。应用新的外显子测序技术发现,该基因突变是米勒综合征——一种经典遗传病,表现为面不和肢体畸形——的罪魁祸首。可见,对人基因疾病相关数据的分析有助于靶点相关副作用的早期发现。
在Jackson实验室的数据库中可以容易地找到已知的表型。为避免纯合敲除小鼠的胚胎致死性,也可以考虑限制性的和/或诱导型敲除小鼠。为评价突变型小鼠的表型,German Mouse Clinic(GMC)已经建立起Helmholtz Zentrum München表型平台,对科研团体开发权限。对于那些作用很强或者不可逆抑制的药物,敲除小鼠的表型学研究对于评估靶点相关的副作用很有帮助。
当已知靶点考虑其他适应症时,已有临床数据的详细研究会很有帮助。医疗靶点数据库(TTD)已经被开发出来提供了很多医疗靶点和相应药物的信息。
从实验室到临床以及其他
靶点验证的主要不足在于针对特定疾病的靶点的有效性只能通过临床试验来判断。如上所述,2008至2010,仅有18%的新药或药物新适应症的II期临床实验取得成功,而失败案例中超过50%都是因为药效不足。从这一角度来看,早期需要考虑验证作用模式的mechanistic biomarkers作为靶点验证的重要关切:
如果mechanistic biomarker显示药物不足以抑制靶点,最好还是寻找更为合适的先导结构。不过,如果mechanistic biomarker证实药物对靶点/通路有很明显的调节作用,但是临床受益却不显著,就要考察该靶点/通路是否有效了。一个典型的案例是Mechanistic biomarker应用于判断血管紧张素II受体拮抗剂用于高血压治疗的开发:I期临床实验中,在健康受试者身上没有发现血压降低效应,但是却检测到了血浆肾素的代偿性增加,这说明该通路在人体内仍然是有效应的。那么,II期临床发现该化合物因为靶点在人体内无效而导致达不到预期临床目标的风险就大大降低了。所以,我们建议尽早使用mechanistic biomarker验证靶点,最好在临床前动物实验中就使用。更多biomarker信息见文献[50]。
所有体内靶点验证的方法,只要可能,都要涵盖已有的工具化合物或者已经建立的药物处方。例如,一个抗肿瘤的新药靶点,不仅需要验证其对转基因/敲除小鼠肿瘤重量的效应,还要评估不进行药物处理时该效应是否还在。这是临床上靶点调节剂面临的一个情况。
总结
本文从拜耳医疗从业科学家的角度讲述了靶点选择标准、靶点验证方法的关键点。不过,就不同的小型、中型和大型公司而言,验证、临床、未被满足的需求、竞争和知识产权等方面在靶点选择的占比会差异很大。
我们相信一个全面的靶点验证会大大降低药物研发后期的失败率。从这个角度来看,如果最初的疾病前体无效,针对该靶点的SMOL或BIOL再好也是没有意义的。