出于时代的自觉,我主动承担了STAMP(Statistical Analysis of Metagenomic Profiles)使用文档的翻译工作。原文28页,翻译如下,欢迎批评。
为了更好的演示,我们先把GitHub上STAMP的文档以及示例文件下载下来。
进入到我的测试目录下,输入如下命令。很神奇,我怎么会用Git?参见第一章《Flask Web开发》:安装flask
$git clone [email protected]:dparks1134/STAMP.git
Initialized empty Git repository in /root/zhouyunlai/fqQuality/MicroFun/STAMP/STAMP/.git/
remote: Counting objects: 1272, done.
Write failed: Broken pipe124/1272), 380.01 KiB 1 KiB/s
fatal: The remote end hung up unexpectedly
fatal: early EOF
fatal: index-pack failed
然而并没有成功,当然你也可以在GitHub直接下载压缩好的文件。出现上面的报错可能是缓存太小,服务器长时间链接无操作导致的。
$vi /etc/ssh/ssh_config
后在文件中添加:
Host *
ServerAliveInterval 120
再clone就可以了。
1. 介绍
STAMP是分析宏基因组(和其他!)概况的软件包,例如
- 物种注释(不同层级的物种丰度)
- 功能注释(不同代谢通路的基因丰度)
它是旨在使用效应大小(effect sizes)和置信区间(confidence intervals)来评估生物学重要性并以此来选择统计检验方法和“最佳实践”的可视化结果。用户友好的图形界面可以轻松探索统计结果和生成出版物级别的统计图,以推断宏基因组(扩增子)中特征(物种和功能)的生物相关性。STAMP是开源的,可通过插件框架进行扩展,并可用于所有主要平台。
2. 联系信息
STAMP正在积极开发中,我们有兴趣讨论这方面的所有潜在应用软件。 我们鼓励您向我们发送有关新功能的建议。 建议,意见和错误报告可以发送到Donovan Parks(donovan.parks [at] gmail.com)。 如果报告错误,请提供尽可能多的信息以及导致错误的数据集的简化版本。 这会让我们能够快速解决问题。
3. 引用STAMP及其统计方法
如果在研究中用到了STAMP,请引用:
Parks DH, Tyson GW, Hugenholtz P, Beiko RG (2014). STAMP: statistical analysis of taxonomic
and functional profiles. Bioinformatics, doi: 10.1093/bioinformatics/btu494.
如果在研究中用到了White’s non-parametric t-test ,请引用:
White JR, Nagarajan N, and Pop M. (2009). Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput Biol, 5, e1000352.
其他统计方法的引用文献,见表1,2,3。STAMP原稿描述参见:
Parks DH and Beiko RG (2010). Identifying biologically relevant differences between metagenomic
communities. Bioinformatics, 26, 715-721.
4.安装
4.1
4.2
4.3
4.4
5.获取和构建宏基因组数据
5.1创建您自己的宏基因组图谱
STAMP的输入文件是以制表符分割格式的(tab-separated values )。文件包含注释层级和样本信息,第一行是每一列的表头,注释信息的列应是自最高层级到最低层级依次排列的。层次结构可以是多层次的,但必须形成严格的树结构。从版本2.0.8开始,STAMP明确检查一个配置文件形成一个严格的层次结构,以确保所有统计测试的有效性。 不幸的是,许多分级分类系统,包括流行的分类法,由于标签错误和其他不一致,如GreenGenes和SILVA目前不是严格等级的。 检查脚本checkHierarchy.py可以从STAMP网站下载并用于识别STAMP配置文件中的所有非层级条目。
必须为每个样本指定在分类层级注释的丰度。在层次结构中的任何点上具有未知分类的读取器应标记为unclassified (不区分大小写),然后可以在多个不同的处理中进行处理 方法(见第6.1节)。 为了允许不同的标准化方法,这些读取计数可以是整数或任何实数。 示例输入文件如下所示:

还必须对分级的子类的父类进行分类。 这可能会导致某些层次结构出现问题,例如GreenGenes和SILVA,其中序列分配给指定的层次结构OTUs,但中间分类学级别通常是未分类的。 为了解决这个问题,我建议从STAMP配置文件中删除OTU列。 如果某些分析(例如,PCA图)需要这种精确度级别,则可以构建单独的STAMP简档,其仅包含OTU列作为分级结构。
5.2创建一个元数据文件(一般的分组文件)
STAMP允许通过元数据文件定义与每个样本关联的其他数据。 像一个STAMP配置文件中,元数据文件是制表符分隔值(TSV)文件。 该文件的第一列表示每个样品的名称,并应对应于相应STAMP配置文件中的条目。 其他列可以指定与正在考虑的样本相关的任何其他数据。 在STAMP中,可以使用这些附加列来定义可以计算统计数据的组(即,一个或多个配置文件的集合)。 例如,上述示例配置文件的元数据文件可能具有以下结构:

5.3
5.4
5.5
5.6
样本量指南
关于检测不同假设检验的统计显著性所需的样本数量有很多文献报道。 为了以实用的建议对此主题进行出色的介绍,我推荐了Suresh和Chandrashekara的文章(2012)以及Jeremy Miles的文章获得合适的样本量
译者注:软件来衡量样本量对统计效力和精度的影响Power and precision,R package pwr也可以。
我的建议如下:
统计假设检验不需要最小样本量有效,但必须满足检验统计量的假设条件(例如,近似正态分布)。小样本量更有可能违反这些假设条件。小样本量也不太可能具有将统计显着性确定为小的效应量所需的统计功效(statistical power)。有趣的是,学生的原始论文(Student’s original paper )证明了t检验考虑的例子,每组只有4个样本。 在这些例子中,由于基础数据的准确性和精确性以及组间效应大小的大小,4个样本是足够的。
例如,考虑尝试确定美国便士的平均重量是否与澳大利亚50美分的平均重量不同。 我把这两个都带到了附近,我可以向你保证,一分钱的重量远远低于过重的澳大利亚50美分! 换句话说,先验我知道效应的大小很大,并且需要更少的样本来检测统计显着性。 此外,这些硬币是在高精度机器上制造的,我们可以使用高精度的刻度精确测量这些碎片的重量。 因此,需要更少的样本来准确估计这些硬币的平均值,并且这些手段周围的变化很小。 由于这些因素(大效应大小,高度精确和精确的测量以及小的方差),小样本量足以确定这些硬币的平均重量在统计上是不同的。
相反,生物数据是嘈杂的。分类学和代谢谱受到很多变异性的影响。与上述示例不同,这些配置文件的准确性和精确度相对较低。更改用于对序列或底层参考数据库进行分类的方法通常会导致对结果配置文件进行实质性更改。这与改变另一个不准确的比例类似。样品制备也会影响产生的曲线。直观地说,我们预计生物复制产生类似的概况,但我们接受将会有很多变异。我们还经常比较广泛定义的群体,我们预计群体内变异很大,例如健康群体与患病群体的群体概况。直观地说,需要大量的样本来可靠地估计这些条件下一组的平均值和方差。因此,在合理比较这两个群体的平均值之前,每个群体需要更多的样本。所需样本的确切数量取决于这些组之间的效应大小,用于定义统计显着性的期望α水平以及期望的统计效力(参见Jeremy Miles文章)。
评估结果时,还必须考虑效应大小。不论样本量大小,两组之间统计学显着性差异的特征可能在生物学上不相关。当样本量很大时,即使极小的差异也会在统计上显着。然而,由于统计学检验不能解释用于产生分类学或代谢谱的方法中可能存在的系统性偏倚,因此当效应量较小时应谨慎。例如,100名健康患者与100名患者的厚壁细胞的小量增加可能仅仅是健康人体内含有更多硬质菌种的参考数据库的结果。当样本量较小时,报告的p值往往不准确,因为统计假设检验不能说明用于产生分类学和代谢特征的方法的准确性和精确度较差。在这些情况下,我认为'最佳实践'(best practice)是使用p值来确定统计显着特征,然后将这些结果进一步过滤为具有足够大的效应大小( effect size)的结果。我认为,不应该报告统计学上不同的特征,也不要指出差异的效应大小。
7.分析宏基因组图谱
7.1分析多个组
设置统计分析属性:可以通过 File->Load
data...对话框加载输入数据。确保在点击OK继续之前指定配置文件(Enterotypes.profile.spf)和组元数据(Enterotypes.metadata.tsv)文件。在这里,我们将按照Arumugam等人指定的三种类型对数据进行分组。 (2011年)。配置文件通过组图例窗口分配给组。要打开此窗口,请选择View->Group legend。组图例窗口可以留作浮动窗口或停靠在不同的位置(图1)。对于此分析,将窗口停靠在右侧(图1b),并从Group字段组合框中选择Enterotype。这表示我们希望按数据组对数据进行分组。如果您打开文件Enterotypes.metadata.tsv,您可以看到Enterotype只是该文件中的一列。已经定义了大量的肠炎型。重复Arumugam等人的分析。取消选中除Enterotype 1,Enterotype 2和Enterotype 3以外的所有组(图2)。在计算统计量和生成图时,取消选中一个组会导致它被忽略。


统计属性通过属性窗口设置。 默认情况下,此窗口停靠在右侧。 但是,它可以从这个位置分离,并停放在不同的位置,就像Group legend窗口一样。 可以使用视图菜单中的相应条目选择性地显示和隐藏Windows。 “属性”窗口允许您设置许多与执行多个组测试相关的属性。 这些在下面描述(图3):
Parent level: 分配给某个要素的序列比例将根据分配给其父类别的序列总数进行计算。 默认值是计算相对于样本中所有已分配序列的比例。 对于本教程,请将父级别保持为整个示例的默认值。
Profile level:构建配置文件的层次级别。 这允许在层次结构中的不同深度探索数据。 对于本教程,将配置文件级别更改为Genera。
Unclassified: 指定未分类序列如何处理。任何分配给名称未分类(不区分大小写)的功能的读取都将被视为Unclassified。Unclassified的序列可以保留在配置文件中(保留未分类的读取),从配置文件中移除(Removeunclassified reads),或者除计算配置文件(仅用于计算频率配置文件)外,不考虑。处理未分类序列的这三个选项可能会导致很大的差异。为保留Unclassified的读取并仅用于计算频率曲线选项,分配给特征的序列的相对比例与指定的父类别内的序列总数成比例。后一个选项可防止Unclassified的功能出现在表格和图表中。相比之下,Removeunclassified reads选项会导致概要文件指示每个特征中序列的相对比例相对于在指定概要文件级别分类的序列。由于样本之间Unclassified序列的比例可能会有很大差异,这可能会导致截然不同的分类。
整体错误率(Family-wise error rate)控制
Statistical properties:统计学测试,post-hoc测试以及置信区间宽度,效应大小和多重测试校正方法(multiple test correction method )的使用都可以在其中指定。
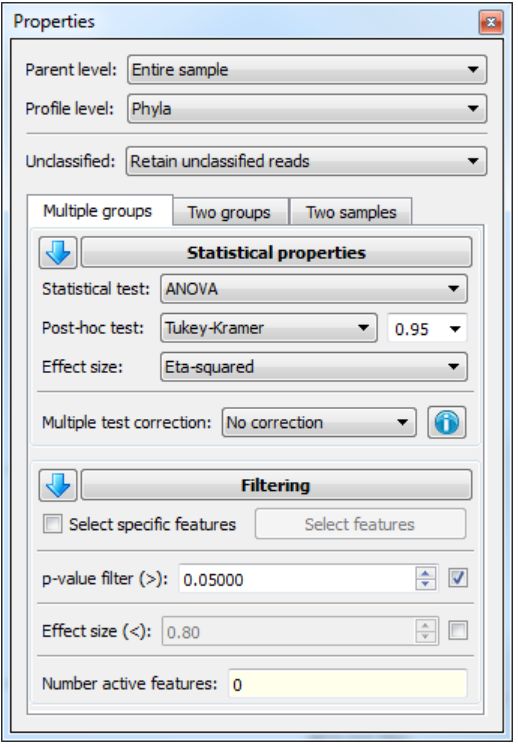
Filtering: 过滤部分提供多个过滤器,用于识别满足一组标准的特征(即,期望的p值和效果大小)。 本节底部显示了通过指定过滤器的功能的数量。 为了允许调查特定功能,STAMP还支持选择功能的子集。 使用Select features对话框执行特征选择,通过单击Select features按钮可以访问该对话框。 在这个对话框中,可以选择或删除特定父类别中的个别特征或所有特征。对这些选定特征执行过滤,以允许调查具有特定特性的特定子集。 要调查一部分功能而不执行任何过滤,请检查所有过滤器。
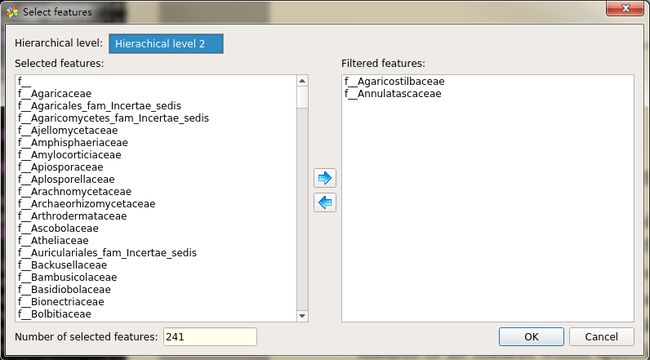
表1中给出了STAMP中用于分析多个组的方法列表。
| 统计假设检验 | 描述 | 参考文献 |
|---|---|---|
| ANOVA | 方差分析(ANOVA)是一种测试几组手段是否相等的方法。 它可以被看作是对两个以上小组的t检验的推广。 | Bluman, 2007 |
| Kruskal-Wallis H-test | 用于测试几组的中位数是否相等的非参数方法。 它考虑每个样本的排序,而不是与某个要素相关的序列的实际比例。 这有利于不假设数据是正态分布的。 每个组必须包含至少5个样本才能应用此测试。 | Bluman, 2007 |
| Post-hoc tests | ↓↓↓↓↓↓↓↓↓↓↓↓↓↓↓↓↓↓↓↓↓↓↓ | ↓↓↓↓↓ |
| Games-Howell | 当ANOVA产生显着的p值时,用于确定哪些方法显着不同。 这个事后测试是为差异和组大小不相等而设计的。 Tukey-Kramer更倾向于当方差不等并且组大小很小时,但它在计算上更昂贵。 | |
| Scheffè | 考虑所有可能的对比的一般事后检验不同于Tukey-Kramer方法,它只考虑一对平均值。 目前,STAMP只考虑一对手段,所以TukeyKramer方法是首选。 一般来说,这个测试非常保守。 | Bluman, 2007 |
| Tukey-Kramer | 当ANOVA产生显着的p值时,用于确定哪些方法显着不同。 它考虑了所有可能的手段对,同时控制了家族错误率(即考虑多重比较)。 一般来说,我们建议在报告最终结果时使用Games-Howell post-hoc测试,并采用Tukey-Kramer方法进行探索性分析,因为它的计算量较小。 Tukey-Kramer也可能是首选,因为它在研究人员中被更广泛地使用和知晓。 | Bluman, 2007 |
| Welch’s (uncorrected) | 简单地对每种可能的手段进行韦尔奇的t检验。 没有努力来控制家族错误率。 | |
| Multiple test correction methods | ||
| Benjamini-Hochberg FDR | 控制错误发现率而不是家族错误的初始建议。 降压程序。 | Benjamini and Hochberg,1995 |
| Bonferroni | 用于控制家族错误的经典方法。 常被批评为过于保守。 | Adbi, 2007 |
| Šidák | 较不常见的控制家族错误率的方法。 一致性比Bonferroni更强大,但需要假设个别测试是独立的。 | Adbi, 2007 |
| Storey’s FDR | 最近用来控制错误发现率的方法。 比Benjamini-Hochberg方法更强大。 需要估算某些参数,并且比Benjamini-Hochberg方法计算量更大。 | Storey and Tibshirani, 2003 Storey et al., 2004 |
表1. STAMP中可用的多组统计技术。 我们的建议以粗体显示.
结果的图形化探索:以下图表用于探索多组分析的结果:
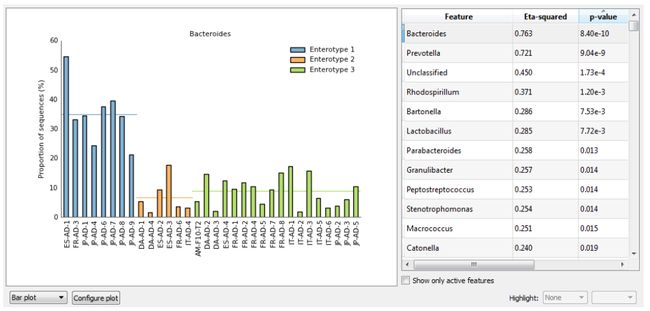
- Bar plot: 条形图指示分配给每个特征的序列的比例。 绘图特征从绘图右侧的表格中选择(图3)。 该表格可以移入和移出以提供剧情的额外空间。 可以对表格列进行排序,以专注于具有较低p值或较大效应大小的特征。 此外,通过选中仅显示活动功能复选框,表格可以限制为通过指定过滤器的功能。 图3中的例子显示了每个样品中拟杆菌属的比例,并揭示了肠杆菌属1内该属的过量丰度。Arumugam et al。 (2011)也建议前列腺球菌和瘤胃球菌属可用于区分肠型。
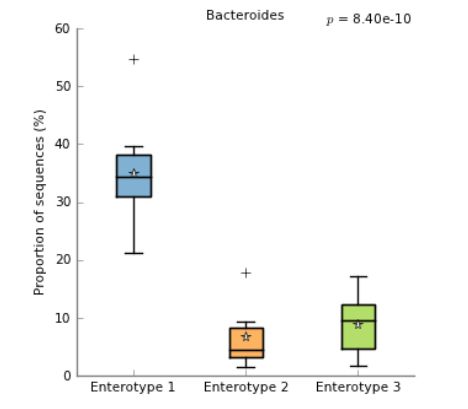
- Box plot 箱形图与条形图相似,除了使用箱须图形指示的组合比例内的比例分布(图4)。 这提供了一个更简洁的比例在一个组内的比例分布。 盒须图形显示数据的中位数为一条线,数据的均值为星号,数据的第25和第75百分位为盒子的顶部和底部,并使用晶须指示最极端的数据 指数中位数为1.5 *(第75百分位 - 第25百分位数)。 晶须外的数据点显示为十字。
- Heatmap plot 指示分配给每个特征的序列比例的热图。 树状图可以沿着热图的两侧显示,并用于聚类特征和样本。 仅绘制活动特征复选框可用于将热图限制为仅通过过滤标准的特征。
- PCA plot: 样品的主成分分析(PCA)图。 点击图中的标记表示由标记表示的样本。 在确定PCA转换之前,除了对数据进行居中之外,没有任何缩放。
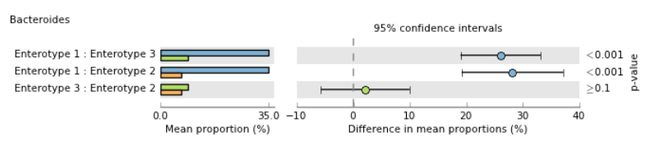
- Post-hoc plot: 多组统计检验(即ANOVA或KruskalWallis)的无效假设是所有组的平均值相等。 如果p值足够小以表明这个零假设应该被拒绝,那么我们只能得出结论,所有组的手段都是不相等的。 如果我们希望确定哪一组对可能彼此不同,则必须进行事后检验。 事后图显示了这种测试的结果。 它为每组对提供了一个p值和一个影响大小的度量(图5)。 在类杆菌的情况下,发现Enterotype 1的平均丰度与2型和3型的平均丰度显着不同(p≤0.001)。相反,肠型2和3的平均丰度没有显着差异(p≥0.1)。
结果表格视图:
多组分析的结果在多组统计表中列表。 该表格通过View-> Multiple group statistics tablemenu项目进行访问。 结果表可以停靠或留作浮动窗口。 可以对列进行排序以帮助识别感兴趣的模式。 通过选中“Show only active features”复选框,可以将结果限制为仅活动特征(通过指定过滤器的特征)。 可以使用Save按钮保存表格。 表格以文本文件形式保存为制表符分隔值格式,任何文本编辑器和大多数电子表格程序都可以读取它们。
7.2 两组间的差异检验
设置统计分析属性:
要分析一组组,请在Properties窗口中单击Two groups选项卡。 无论是分析多个组还是一对组,分组均由Group legend窗口中Group字段组合框的值确定。 在本节中,我们将考虑通过将分组字段设置为性别,男性和女性的肠道微生物群是否存在组成差异。
sed ":a;N;s/\n//g;ta" ll # sed去掉换行符
统计属性通过属性窗口设置。 父级别,简档级别和未分类序列的处理设置均适用于所有分析(即多个组,两个组和两个样本)。 分析特定属性在“属性”窗口的分析类型选项卡下给出。
Profile:配置文件部分用于指定将分析哪组配对。 在这种情况下,我们只有两个组(男性和女性),所以我们不需要改变这些值。 通过点击这些组旁边的颜色按钮,也可以更改与两组相关的颜色。 组2也可以设置为<所有其他样本>,在这种情况下,所有不包含组1的样本都用于组成第二组。 这对于将一组特定的样本与研究中的其他样本进行比较是有用的。
Statistical properties :统计测试,置信区间方法和宽度,以及多个使用的测试校正方法都可以在本节中指定。 可以进行单侧或双侧的统计假设检验,尽管通常应该使用双侧检验来解决在Rals等人讨论的原因。(2007年)。 表2给出了STAMP中分析两组的方法列表。
| 统计假设检验 | 描述 | 参考文献 |
|---|---|---|
| t-test (equal variance) | 明确假定两组具有相同方差的学生t检验。 当这个假设可以做出时,这个测试比Welch的t检验更强大 | Bluman, 2007 |
| Welch’s t-test | 当两组不能被假定具有相同的方差时,用于学生t检验的变体。 | Bluman, 2007 |
| White’s non-parametric t-test | White等人提出的非参数测试。 用于临床统计学数据。 该测试使用排列过程来消除标准t检验的正态性假设。 此外,它使用一种启发式方法来识别稀疏特征,这些稀疏特征由Fisher的精确测试(Fisher’s exact test)处理,并且当任何一个组由少于8个样本组成时,用池化策略处理(a pooling strategy)。 有关详细信息,请参见White et al。,2009。对于大型数据集,此测试可能在计算上很昂贵。 它可能有助于减少可在Preferences-> Settings对话框中设置的复制次数。 | White et al., 2009 |
| 置信区间方法 | ||
| DP: t-test inverted | 只有在使用相等方差t检验时才可用。 通过反转等方差t检验来提供置信区间。 | |
| DP: Welch’s inverted | 仅在使用Welch的t检验时才可用。 通过反转韦尔奇的t检验来提供置信区间。 | |
| DP: bootstrap | 仅在使用White的非参数t检验时才可用。 使用百分比自举方法提供置信区间。 如果White的非参数t检验默认使用Fisher精确检验,则使用CC渐近方法获得置信区间(参见表3)。 | |
| 多重检验校正方法 | ||
| Benjamini-Hochberg FDR | 控制错误发现率而不是家族错误的初始建议。 降压程序 | Benjamini and Hochberg, |
| 1995 | ||
| Bonferroni | 用于控制家族错误的经典方法。 常被批评为过于保守。 | Adbi, 2007 |
| Šidák | 较不常见的控制家族错误率的方法。 一致性比Bonferroni更强大,但需要假设个别测试是独立的。 | Adbi, 2007 |
| Storey’s FDR | 最近用来控制错误发现率的方法。 比Benjamini-Hochberg方法更强大。 需要估算某些参数,并且比Benjamini-Hochberg方法计算量更大。 | Storey and Tibshirani, 2003 Storey et al., 2004 |
表2. STAMP中可用的两组统计技术。 我们的建议以粗体显示。 DP =平均比例之间的差异。
两组间的比较为什么还要做多重检验分析?因为每个样本有多个指标。高通量数据的多重检验问题
Filtering:过滤部分提供了大量的过滤器,用于识别满足一套标准的特征,其中特征的数量通过在该部分底部指明的指定过滤器。注意可以使用选择特征来关注特定的特征子集 对话。 提供的过滤器如下所示:

- p-value filter: 删除p值大于指定值的所有要素
- Sequence filter: 允许删除已分配少于指定数量序列的功能。 对于给定特征的序列的最大或最小数量,可以将过滤应用于两组中的样本。 或者,可以独立地应用过滤到每个组内的样本,并且如果任一组内的样本含有不足数量的序列,则过滤特征。
- Parent sequence filter: 与序列过滤器相同,除了应用于父类别中的序列计数。
- Effect size filters: 效果大小过滤器:可以移除小效果大小的功能。 可以在两个不同的效果大小统计信息上执行过滤。 这允许人们既过滤绝对(即,比例之间的差异)和有效化的相对(即比例比率)度量。 可以应用这些过滤器,以便过滤掉任一条件(逻辑OR运算符)或两个条件(逻辑AND运算符)的功能。 这些效应大小过滤器适用于组内所有样本的平均比例。
结果的图形化探索:
提供以下图表用于探索两组分析的结果:
- Bar plot :指示分配给每个特征的序列比例的条形图。从图表右侧的表格中选择要素图的特征。
- Box plot:箱形图类似于条形图,除了使用箱须图形指示的组合比例内的比例分布。这提供了一个组中比例分布的更简洁的总结。盒须图形显示数据的中位数为一条直线,数据的均值为星号,数据的第25和第75百分位为盒子的底端,并使用晶须指示最极端的数据在中位数的1.5 *(第75百分位 - 第25百分位)内。晶须外部的数据点显示为交叉点。
- Heatmap plot:指示分配给每个特征的序列比例的热图。可以在热图两侧显示聚类图,并用于聚类特征和样本。仅绘制活动特征复选框可用于将热图限制为仅通过过滤标准的特征。
- PCA plot :样本的主成分分析(PCA)图。点击图中的标记指示由标记表示的样本。在确定PCA变换之前,除了对数据进行居中之外,不进行任何比例缩放。
- Scatter plot :指示每个组中分配给每个特征的序列的平均比例。这个图对于识别明显富集于其中一个双组中的特征非常有用。每个组内数据的扩散可以用不同的方式显示(例如,标准偏差,最小和最大比例)
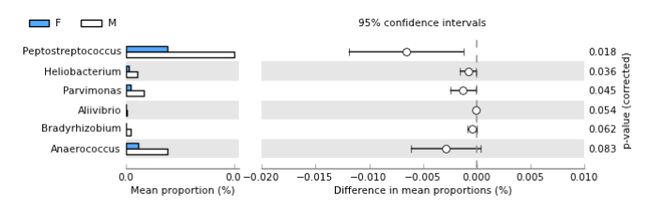
- Extended error bar: :表示两组之间平均比例的差异以及该效应大小的相关置信区间和指定统计检验的p值。另外,条形图指示在每组中分配给一个特征的序列的平均比例。我们认为这是推断特征生物相关性所需的最少量信息。图6给出了肠炎菌型数据的扩展误差棒图。
结果表格视图:两组分析的结果列在两组统计表中。 该表格通过View->Two group statistics table菜单项访问。
7.3 两个样本之间的差异检验
设置统计检验参数
要分析一对样本,请单击属性窗口中的两个样本选项卡。 在本节中,我们将考虑两个双胞胎AM-F10-T1和AM-F10-T2之间肠道微生物群是否存在组成差异。
