说明:此篇笔记系2016-2017年由克里克学院与康昱盛主办的蛋白质组学网络大课堂整理而成,侵删。该课程由上海交通大学系统生物医学研究院助理研究员库鑫博士所授。
主要知识点:
- 基于质谱的蛋白质组学:质谱技术的前世今生
- 蛋白定性检测:背景介绍、二级质谱、鉴定碎片离子
基于质谱的蛋白质组学
大伙儿都知道,蛋白质组学(proteomics),是研究一种细胞或者一种生物体所表达的全部蛋白质。虽说现在基因组测序火得一塌糊涂,但是,我们不要忽略了,蛋白质才是执行生命体功能的基本单元,而且蛋白质都是通过形成各种复合物,组成通路网络,去行使各种生物学功能的!所以,有很多生物学问题只能在蛋白质层面上去研究去探索,而且需要站在系统的层面去考察,比如说:蛋白-蛋白相互作用、蛋白的细胞定位、翻译后修饰、信号通路及代谢通路的调控和功能等。这就是为啥蛋白质组学如此重要啦!
既然重要,科学家们自然是想尽办法来研究了!最开始使用的技术就是传说中的双向凝胶电泳(2-DE),由于分辨率低、蛋白质重叠等各种问题,无论是通量还是准确度,都不尽如人意。当质谱技术兴起以后,就迅速被替代了。

说起质谱技术的诞生,估计很多小伙伴都听过那个著名的diao丝逆袭的段子,讲的就是2002年诺贝尔化学奖得主田中耕一,作为蛋白质谱发明人之一,由于一个不小心在实验时错加了甘油,结果神奇地将质谱技术引入到鉴定生物大分子的应用领域。想想,大到整个人类的科技发展史,小到每个个体的人生,都充满了多少不可思议~
当质谱技术与蛋白质组学碰到了一起,真是天雷引了地火,产生出强烈的化学反应,迅速引爆整个学科的发展!也就十几年的时间吧,蛋白质组学的研究目标从细胞模型、动物模型,到人的体液、组织等人体样本,应用范围的生物复杂度越来越高。研究目的呢,也从最初的肽段序列推导,到多肽和蛋白质的定性定量分析,翻译后修饰,再到如今成为新热点的靶向蛋白质组学,总之,势不可挡啊!
说到靶向蛋白质组学,咱们都知道,一直以来蛋白质组学的应用领域主要是针对基础生物学,比如研究通路、蛋白复合物、互作网络,表征细胞和组织的类型,观察细胞周期内蛋白质的表达等。近年来,由于技术的飞速发展,蛋白质组学开始被用于医学研究和药物研究。比如说药物研究,国内可能用得还不多,但在欧美已经开始越来越广泛。以肝毒性为例,蛋白质组学可以为药物研发前期的肝毒性评估提供研究手段。
那么,怎么将蛋白质组学应用到临床及药物研发中呢?就是需要靶向蛋白质组学技术了!以前,蛋白质组学技术主要用于发现新的未知物,比如肽段、蛋白复合物、蛋白的翻译后修饰等。这部分的应用很广,技术门槛比较低,方法比较通用。但问题是,这种方法思路没办法应对大量的临床样本,可重复性和准确性达不到要求。
于是,靶向分析开始兴起,就是说,分析之前我们就明确知道需要分析的物质是什么,然后把它挑出来,进行一个精确的定量和分析!我们不需要一次性验证成千上万的蛋白,但我们需要在成百上午的样本中验证十几种或者几十种我们关心的蛋白质,而且这些蛋白质常常都是浓度很低的蛋白,用传统的方法基本上只有被遗漏的命(后面小编会详细讲为什么会遗漏)。有了靶向技术,对于研究临床诊断的生物标志物,就有了更大的可能和更强的支撑了!

那么接下来,根据老师讲课的思路,小编就从定性检测、定量检测和靶向蛋白质组学三个方面来分享下听课的收获。
定性检测
无论是定性还是定量检测,样品制备是跑不掉的准备工作。用于质谱的蛋白质样品,来源非常广泛,只要你是包含了蛋白质的东西,都可以作为来源。对于复杂的样品,比如人体体液或组织样本,蛋白质的提取及去高峰度,常常需要复杂的精细的处理,而且处理流程根据样本和研究目的的不同而不同。这部分内容呢,第二讲“样品前处理”会详扒,感兴趣的小伙伴可以期待小编的下一篇听课笔记吧~
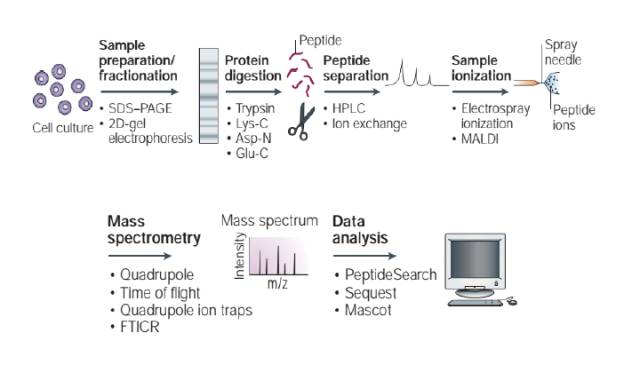
话说,蛋白质的定性检测有两种思路:Bottom-up和Top down。Top down是指从一个完整的蛋白出发,在质谱中进行碎片化处理,通过对碎片分子的检测,推导出蛋白的序列。而在使用中真正占绝大多数是Bottom-up方法,也就是我们常说的shotgun方法,它充分利用了蛋白质自身的特点:可以被特定的酶在特定的位点切断。基本思路是,先用蛋白酶把蛋白序列进行酶切,再针对酶切后的肽段进行鉴定,所以进入质谱的检测对象永远是肽段,再根据肽段序列再推导出蛋白序列。

1. 样本处理:拿到蛋白来源的各种样本,进行前处理和优化。
2. 蛋白分离:根据研究需要,用凝胶分离,提取所需的蛋白,或者不分离,全部拿来检测,需要注意去杂质;
3. 酶切:用序列特异性的酶,对蛋白进行酶切;
4. 肽段分离:酶切后的肽段进入HPLC(高压液相色谱),这也就是我们常说的LC-MS中的LC,肽段会因为在色谱柱填料上的保留时间的不同,得到预分离;
5. 电离:分离后的肽段,加电压使其离子化(ESI);或者用MALDI基质辅助的激光解离,就不需要HPLC的过程;
6. 质谱解析:将带上电荷的肽段送入质谱,肽段会在磁场中发生偏转(质谱仪的基本原理),在质谱里收集信号,得到谱图。
7. 搜库:用搜索软件对质谱图进行自动化的分析,得到肽段及蛋白序列信息。
Tips
质谱种类很多,比如四级杆质谱、飞行时间质谱、四级杆离子阱、傅里叶变换质谱等。
换个角度,对Shotgun方法的流程,我们可以这样来总结:
数据产生:蛋白->肽段->谱图
数据分析:谱图->肽段->蛋白
这里面最关键的一个指标,我们叫Peptide-Spectrum matching(PSM),就是指谱图与肽段的匹配。匹配得越好,则反推出的蛋白就越准确。这个匹配的过程,也就是我们常说的搜库。那么接下来小编就来分享一下从课程中学习到的搜库背景知识、搜库工具和算法,以及对搜索结果的评估。
背景介绍
质谱,听上去很高大上,无论有多贵重,都是由三部分组成的:离子源+质量分析器+检测器。

一台质谱可以不止一个离子源\分析器\检测器,可以把几种串联起来,针对不同分析需要来使用。
离子源
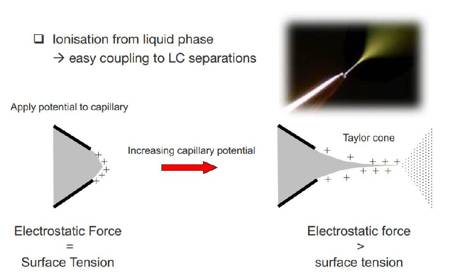
我们先来说说离子源。蛋白质谱所使用的ESI(Electrospray ionization)电喷雾离子化,对蛋白质组学来说是一个标志性的发明!因为是直接从液相进行离子化,使它与LC(液相色谱)的联用变得更加容易了,我们可以先用LC将非常复杂的肽段混合物进行预分离,减少每次分析物的复杂度,然后分离的肽段可以直接进入ESI,形成电离喷雾。
那么,ESI喷雾是怎么形成的呢?简单来说,分离柱前端有一个小开口,被分析物根据质量及电荷的不同,依次通过前端的小开口。小开口处加了电压,刚开始,静电力与表面张力相同,当加大静电力使它大于表面张力的时候,液膜破裂,形成无数带电的小液滴,就形成喷雾了。像现在比较新的nanoESI技术,LC的流速就更加慢,离子化的效果也更好。觉得以上描述还不够形象的童鞋,直接看图吧:
质量分析器
说完了离子源,接下来我们来说质量分析器,这是质谱仪里最重要的一部分。我们通常听到的各种质谱仪的名字,就是根据质量分析器的类型来命名的。我们样品中各组分在离子源中发生电离,并经加速电场的作用后,形成离子束,进入质量分析器中。质量分析器将带电离子根据其质荷比加以分离,记录各种离子的质量数和丰度,用于后续定性与定量的分析。
质量分析器有两个主要的技术参数:质量范围和分辨率。质量范围是指是所能测定的质荷比的范围,它决定了咱们能检测到的离子的范围。比如,ESI离子源能产生许多m/z大于3000的离子,如果你选的质量分析器的上限达不到3000,那么3000以上的离子你就检测不出来了。
然而,另一个更为重要的指标,就是质量分析器的分辨率!先上个公式描述:
分辨率=观测的一个质谱峰的质荷比/半峰高处的峰宽(FWHM)
啥意思呢?比如下图中最左边的那个峰,它的质荷比是1,085.55,峰高一半的地方的峰宽值是0.217,于是:
分辨率=1,085.55/0.217=5,000

如果这么讲还是不太明白,那你可以简单理解为,质谱分辨率越高,我们将得到越尖越细的谱峰。你可能会问:谱峰又尖又细的好处是什么?这是个好问题!事实上,分辨率可以表征两个相邻的谱峰在质谱中被区分开的能力。大家通过下图感受一下不同分辨率的质谱仪能给我们多么不同的谱峰图。
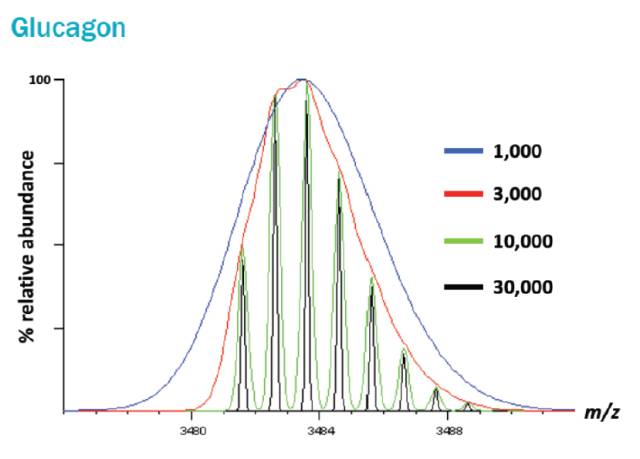
图中以Glucagon(胰高血糖素)为例,展示了不同分辨率的质谱仪给出的谱峰。当分辨率是1000时,只能看一个很宽的峰(蓝色);分辨率增加到3000时,峰窄一些(红色),但还感受不到明显的差别;当提高到10000时,很明显能看到,其实这里包含了8个峰(绿色);再提高到30000的时候,半峰宽更窄,两个相邻的峰可以彻底地被分开(黑色)。显然,我们在分辨率为1000或3000,不能准确的检测被分析肽段的精确分子量, 从而导致谱图无法匹配或者发生错配。
不同的质量分析器有不同的分辨率,通常的顺序是:傅里叶变换质谱分辨率最高,但造价太贵;其次是Orbitrap(轨道阱系列),分辨率远远高于其它质谱;再次是TOF(时间飞行质谱);然后是离子阱(Ion Trap);最后是四级杆质谱(Quadrupole)。
这里小编多说一句,分辨率高固然好,但价格肯定就贵,选择质谱仪的时候要根据咱们自己的研究目的以及预算范围啦!
二级质谱
然而,要对肽段进行鉴定,一级质谱显然是办不到的,我们没法根据肽段离子m/z的值就推断出这个肽段由哪些氨基酸残基组成(可能的组合非常多),以及序列顺序是怎么样的,对吧?所以,鉴定肽段还需要二级质谱。
什么是二级质谱呢?简单来说,肽段混合物通过一级质谱得到了一级谱图,然后从中选择一个肽段,通过一些方法,比如,与随性气体进行碰撞,把肽段碰碎,得到碎片离子,再形成二级谱图。我们通过观察碎片离子的质量分布来推断肽断的残基组成,最后再反推出蛋白质是什么。上个图,帮助大家理解一下二级质谱是怎么来的。
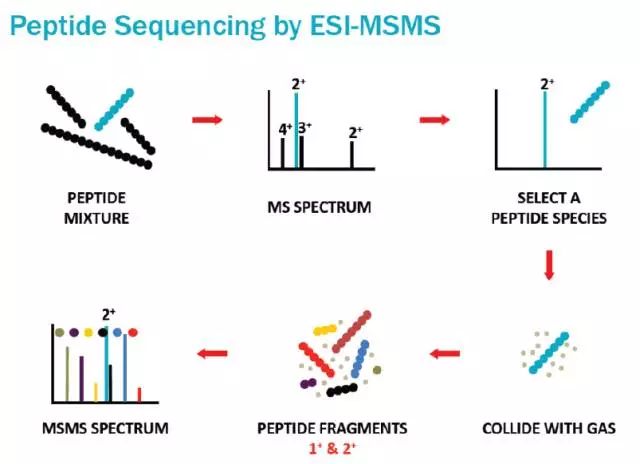
在上一段,小编提到是从一级质谱中“选择”一个肽段进入二级质谱。这里看似讲得云淡风轻,事实上怎么选却是一个很关键的问题!通常选择的方法我们可以叫做“TOP”法(这是小编自己起的名字),比如TOP15就是指从一级谱里选前15个高度的峰,每一次分离一个肽段,然后对这个肽段进行扫描,得到二级谱图。
大家发现了没有?如果一个肽段在一级谱图中没有进入TOP15,那它连打二级谱图的资格都没有!原来质谱的世界竞争也是如何残酷!二级质谱能扫描哪些肽段是由一级质谱决定的,所以我们将这种方法称为“数据依赖性采集(DDA, data dependent acquisition)!
明白了吧,DDA这个名字就是这么来的!下次大伙儿再听到有人说DDA,心里不会再一百个问号飞过了吧?
咱们细想一下就不难发现,如果一个蛋白的浓度不够高,也就是说,它的肽段在一级谱图中很难成为那些TOPs,那么它能进入二级质谱的可能性基本上没有。这就是为什么低峰度蛋白很难被鉴定到!这也就是为什么我们在做比如血液这种样品的时候,一定要去除血红蛋白等高峰度蛋白(如果你想鉴定的蛋白不是血红蛋白的话)!
很显然,DDA方法的局限性就摆在那里!这叫想要研究低峰度蛋白的科学家们怎么忍?于是,一种叫做数据非依赖性采集(DIA)的新方法就应运而生了!关于这种方法的原理,下一篇推文会详扒。
我们再通过以下这个图来感受一下一级谱图与二级谱图之间的关系:
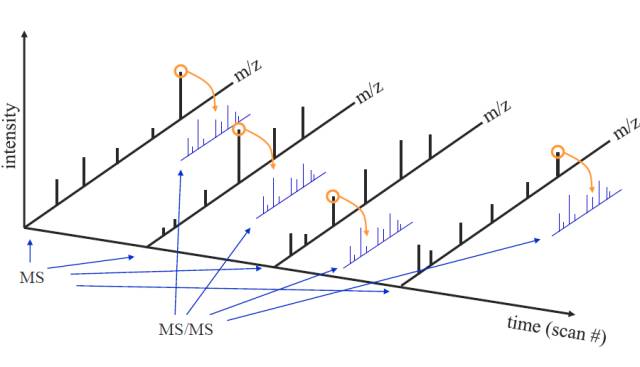
比如,第一个时间点,我们先进行MS1扫描,然后选一个峰高的肽段进行MS2扫描,依次类推。在一些扫描速度比较快的质谱仪里,一个MS1谱图可以进行80张MS2的扫描。
鉴定碎片离子
好,我们搞清楚了二级质谱是怎么来的,那么我们怎么根据检测到的离子信息来推测这是什么氨基酸呢?可能你会说,这还不简单么?根据分子量呀!
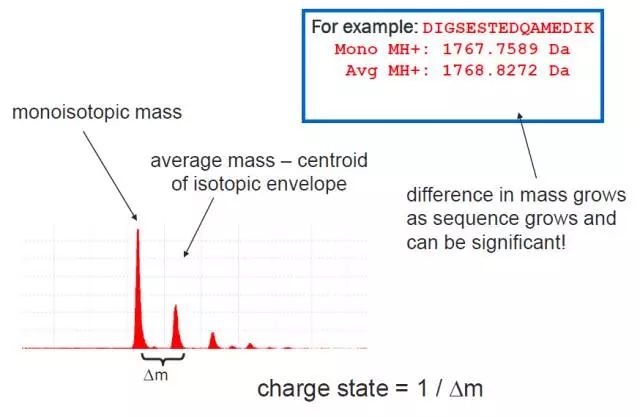
没错,不同的氨基酸,它的分子量不就是一个简单的值吗?然而,这件事却并没有这么简单,因为这个世界上还存在一个神奇的东西,它的名字叫同位素!
比如说碳元素,最常见的是原子量12的这种,我们叫C12,然而它还有一个同样很稳定的好基友,C13(多一个中子)。于是,我们得考虑到这两种稳定同位素的含量(百度百科说C13占 1.11%,C12占98.89%),对于一个氨基酸而言,我们就会得到两个不同的分子量:
单同位素分子量,也就是只包含比例最高的那一种同位素的分子量;
平均分子量,也就是包含了多种同位素的平均分子量。
为啥说平均呢?因为当肽段分子量越大,含有各种同位素的可能性及不同组合就越多,我们如果把每一种组合都算一遍分子量,这样会得到一个长长的list,到时候做谱图匹配时用哪一个值呢?也没谱。所以干脆用一个平均值来表示。
我们通过下表来感受一下各种不同的氨基酸残基的单同位素分子量与平均分子量有多大的区别:
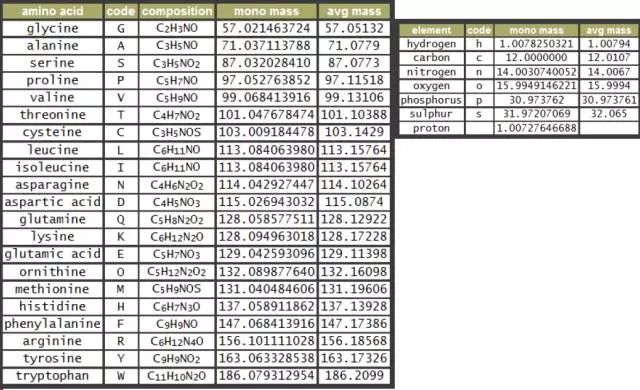
可能你又会问,这两个不同的分子量分别在什么情况下用呢?这里又要说到分辨率了,如果咱们用的是高分辨率质谱仪,不同的同位素峰会被明显地分开,也就是说,谱图里我们能看几个同位素峰,这时我们就可以使用单同位素分子量,可以与相应的单同位素峰准确对应。但在低分辨率质谱仪里,这些峰很可能混在一起,看上去只是一个峰,这种情况下,也没办法,只能用平均分子量去近似一下了。
下面这个图可以很形象地展示出,单同位素分子量与平均分子量在质谱图上差别有多大。在高分辨质谱看来,这完全就是两种不同的离子了。上面我们也说了,根据平均分子量来计算,结果并不准确,但用单同位素分子量来计算,就可以准确对应了。
除了同位素,还有一个因素我们也需要考虑,那就是肽段碎裂进入二级质谱时,可能会形成三种不同的离子类型,这就是我们通常所说的by离子,ax离子和cz离子。

之所以会形成不同的离子对,是因为不同的碎裂方法,造成肽段断裂的位置不同。大伙儿看看上面这个图就明白了。当我们使用CID(碰撞诱导解离)或HCD(High-energy C-trap Dissociation)碎裂时,与惰性气体碰撞的是C-N键这里,C端生成y离子,N端生成b离子,这是二级质谱产生的最常见的离子对了。当我们使用ETD(电子转移解离)碎裂时,因为有一个电子反应的过程,在加上电子后才产生的碎裂,它的断裂位置可能出现在N-C键这里,形成cz离子,而TOF类仪器可能会产生ax离子。
离子类型的信息需要传递给后续的搜库步骤(通常我们在搜库软件中指定了仪器类型,软件就会自动匹配离子类型),计算机需要模拟最可能的碎裂位置,生成对应的理论谱图,然后拿来与实际谱图比对。我们以by离子为例,来看看对一个肽段来说,它可能碎裂成哪些碎片离子:
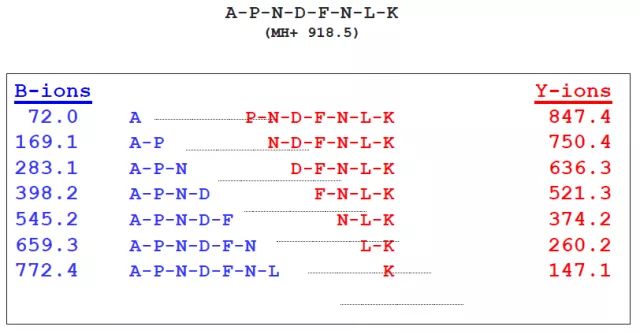
那么它可能会生成如下这样的谱图:

从谱图上看,这个肽段所有的by离子都检测到了。通常来说,对于丰度不错,长短合适的肽段,在高精度质谱仪上被完整捕获到的情况是很常见的。通常情况下50%-80%的by离子都能被捕获到。
下篇继续讲定性检测里的搜库工具、结果评估,以及定量检测的各种背景知识。