概要
本文主要讨论Seurat对象导入到Monocle中直接进行分析的可行性,分两种情况:
①经过数据清洗、标准化和聚类的Seurat对象导入
②未经过任何处理的Seurat对象导入
以下先进行Monocle包的简单介绍,再分这两种情况进行尝试。
为什么要分这两种情况进行尝试?
- Seurat包中也有将数据标准化的步骤,作者的建议是在Monocle中要再次进行标准化,但是他自己也没有尝试过,所以不确定会怎么样。
- Seurat包中有个
ScaleData的命令,目的是去除测序产生的批次效应和技术噪音,但对于我们的数据(按不同时间缺血处理的脾脏,根据锥虫感染小鼠的时间进行测序),我们要观察的就是这些不同时间批次之间的差别,有可能这个命令会将这个差别掩盖了。因此如果直接输入已经聚类好的Seurat对象,也许会出现问题。
关于Monocle
http://cole-trapnell-lab.github.io/monocle-release/
【Introduction】
Monocle介绍了使用RNA-Seq进行单细胞轨迹分析的策略,能够将细胞按照模拟的时间顺序进行排列,显示它们的发展轨迹如细胞分化等生物学过程。Monocle从数据中用无监督或半监督学习获得这个轨迹。
无监督:利用Monocle的自己一套工具或Seurat生成一个基因列表
半监督:通过自身的知识积累人为输入一些认为重要的基因
Monocle不是通过实验将细胞纯化为离散状态,而是使用算法来学习每个细胞必须经历的基因表达变化的序列,作为动态生物过程的一部分。一旦它了解了基因表达变化的整体“轨迹”,Monocle就可以将每个细胞放置在轨迹中的适当位置。然后,可以使用Monocle的差异分析工具包来查找在轨迹过程中受到调控的基因。如果该过程有多个结果,Monocle将重建“分支”轨迹。这些分支对应于细胞“决策”,Monocle提供了强大的工具来识别受其影响的基因并参与制作它们。网站中也提供了分析分支的方法。Monocle依靠Reversed Graph Embedding的机器学习技术来构建单细胞轨迹。
除了构建单细胞轨迹之外,它还能够做差异表达分析和聚类来揭示重要的基因和细胞。这与Seurat的功能相似。
【Workflow以及与Seurat的异同】
①创建CellDataSet对象(下简称CDS对象)
若要创建新的CDS对象,则需要整理出3个输入文件(基因-细胞表达矩阵、细胞-细胞特征注释矩阵、基因-基因特征注释矩阵),但方便的是,Monocle支持从Seurat中直接导入对象,通过importCDS命令实现。
在创建之后,也会进行数据过滤和标准化,不同的是Seurat是基于作图的方法去筛选掉异常的细胞,而Monocle的过滤方法则是根据表达数据的数学关系来实现。
上限:
下限:
其中为基因表达总数, n 为细胞数,为表达水平方差
大概就是以一个大致的细胞表达水平为基准,表达量太高的跟太低的去除掉。②通过已知的Marker基因分类细胞
方法一:通过一些现有的生物/医学知识手动赋予它们细胞名,将这些细胞分为几大类,然后再聚类细胞。
方法二:与Seurat包的分类细胞方法类似,筛选出差异表达基因用于聚类,然后再根据每一个cluster的marker赋予细胞名。③聚类细胞
采用的也是t-SNE算法。-
④将细胞按照伪时间的顺序排列在轨迹上
- Step1:选择输入基因用于机器学习
这个过程称为feature selection(特征选择),这些基因对轨迹的形状有着最重要的影响。我们应该要选择的是最能反映细胞状态的基因。
如果直接通过Seurat输出的一些重要的基因(比如每个cluster中的高FC值基因)作为输入对象的话就能够实现一个“无监督”分析。或者也可以利用生物学知识手动选择一些重要的基因进行“半监督”分析。 - Step2:数据降维
利用Reversed graph embedding算法将数据降维。
相对于PCA来说这个算法更能够反应数据的内部结构(据monocle网站说是这样) - Step3:将细胞按照伪时间排序
这个过程称为manifold learning(流形学习)。Monocle利用轨迹来描述细胞如何从一个状态转换到另一个状态。得到的是一个树状图,树的根部或树干表示的是细胞最初的状态(如果有的话),随着细胞的变化就沿着分叉树枝发展。一个细胞的伪时间值(pseudotime value)为它的位置沿着树枝到根部的距离。
- Step1:选择输入基因用于机器学习
⑤差异表达分析
还没细看
情况①:经过清洗、标准化和聚类的Seurat对象导入
spleen
An object of class seurat in project 10X_spleen
15655 genes across 1940 samples.
clustered_spleen_monocle <- importCDS(spleen, import_all = TRUE)
head(pData(clustered_spleen_monocle))
nGene nUMI orig.ident percent.mito res.0.6 Size_Factor
AAACCTGAGGTGATAT 1072 3999 10X_spleen 0.01150863 3 NA
AAACCTGAGTCATCCA 1562 4640 10X_spleen 0.03709295 5 NA
AAACCTGCAGTGAGTG 995 3489 10X_spleen 0.03955288 3 NA
AAACGGGAGACTGGGT 1704 7397 10X_spleen 0.02852508 0 NA
AAACGGGAGACTTGAA 976 3102 10X_spleen 0.04932302 2 NA
AAACGGGAGATAGGAG 1236 4548 10X_spleen 0.02682498 1 NA
res.0.6为cluster的编号,将此列名更改为“Cluster”,方便后面使用。
names(pData(clustered_spleen_monocle))[names(pData(clustered_spleen_monocle))=="res.0.6"]="Cluster"
range(pData(clustered_spleen_monocle)$Cluster)
[1] "0" "8"
确认此列的范围为0到8,也就是共9个cluster。
计算SizeFactor和Dispersions
计算用于方便之后的分析使用。
clustered_spleen_monocle <- estimateSizeFactors(clustered_spleen_monocle)
clustered_spleen_monocle <- estimateDispersions(clustered_spleen_monocle)
跳过-数据清洗
由于此数据已经经过数据清洗,因此没有必要在Monocle中再一次处理。
数据标准化
根据作者的建议,即使在Seurat包中已经标准化处理过的数据,在转化到Monocle中时仍然需要再一次进行标准化。
#将表达矩阵中所有值进行log标准化
L <- log(exprs(clustered_spleen_monocle[expressed_genes,]))
#将每个基因都标准化,melt方便作图
library(reshape)
melted_dens_df <- melt(Matrix::t(scale(Matrix::t(L))))
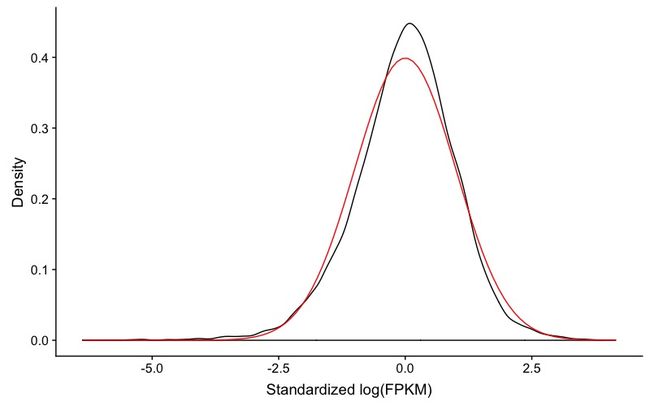
#作图,查看标准化的基因表达值的分布
qplot(value, geom = "density", data = melted_dens_df) +
+ stat_function(fun = dnorm, size = 0.5, color = 'red') +
+ xlab("Standardized log(FPKM)") +
+ ylab("Density")
利用细胞Marker分类细胞
首先,因为一种细胞下又可以细分成更加小的类别,因此在用marker给细胞类时,就要考虑到它们的对应关系。如CD4基因所对应的细胞为CD4+ T细胞,而CD4+T细胞属于T细胞中的一种,我们就要告诉Monocle CD4+T细胞属于T细胞的子集,让它不要再分类的过程中将它们分成两类。
Monocle提供了一个将细胞分级的函数newCellTypeHierarchy.
cth <- newCellTypeHierarchy()
将marker和细胞对应起来,以及排列好它们的从属关系。
cth <- addCellType(cth, "T cell",
classify_func=function(x) {x["CD3D",] > 0})
cth <- addCellType(cth, "CD4+ T cell",
classify_func=function(x) {x["CD4",] > 0},
parent_cell_type_name = "T cell")
cth <- addCellType(cth, "CD8+ T cell",
classify_func=function(x) {
x["CD8A",] > 0 | x["CD8B",] > 0
},
parent_cell_type_name = "T cell")
cth <- addCellType(cth, "B cell",
classify_func=function(x) {x["MS4A1",] > 0})
cth <- addCellType(cth, "Monocyte",
classify_func=function(x) {x["CD14",] > 0})
cth <- addCellType(cth, "Neutrophil",
classify_func=function(x) {x["S100A9",] > 0})
下面这一步将细胞进行分类。
clustered_spleen_monocle <- classifyCells(clustered_spleen_monocle, cth, 0.1)
查看细胞分类的情况。
table(pData(clustered_spleen_monocle)$CellType)

伪时间分析
Step1: 选择定义细胞发展的基因
diff_test_res <- differentialGeneTest(clustered_spleen_monocle,fullModelFormulaStr = "~Cluster")
ordering_genes <- row.names (subset(diff_test_res, qval < 0.01))
Step2: 数据集降维
clustered_spleen_monocle <- setOrderingFilter(clustered_spleen_monocle, ordering_genes)
plot_ordering_genes(clustered_spleen_monocle)
clustered_spleen_monocle <- reduceDimension(clustered_spleen_monocle, max_components = 2, reduction_method = "DDRTree")
Step3: 将细胞按照伪时间排序
clustered_spleen_monocle <- orderCells(clustered_spleen_monocle)
查看能用于上色区分的变量:
colnames(pData(clustered_spleen_monocle))
[1] "nGene" "nUMI" "orig.ident" "percent.mito" "Cluster"
[6] "Size_Factor" "CellType" "Pseudotime" "State"
其实这一步按照正常情况下来说,是应该最好有一个时间的变量(如Hour或者Time),用来区别不同时间批次处理产生的数据,子亮部分的数据就可以根据不同的寄生时间来给细胞上色,从而观察随着寄生时间的变化细胞状态(发育/分化)的变化。而像脾脏数据这一种,虽然按照了4个时间点进行了处理,但是数据并没有按照不同时间点区分出来,因此只能够根据细胞的分化的过程来确定哪些是原始状态。
plot_cell_trajectory(clustered_spleen_monocle, color_by = "Cluster")
plot_cell_trajectory(clustered_spleen_monocle, color_by = "CellType")
plot_cell_trajectory(clustered_spleen_monocle, color_by = "State")
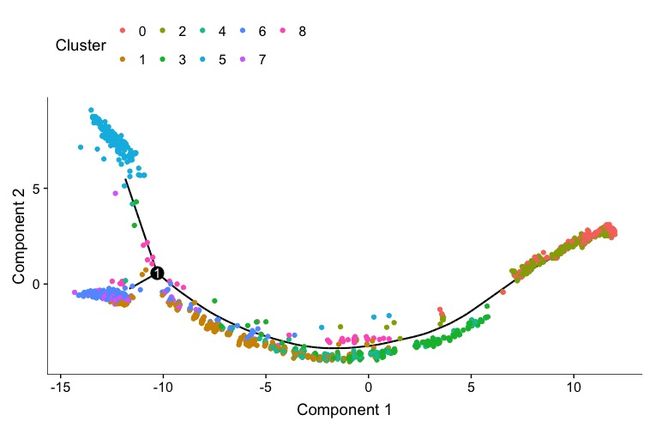
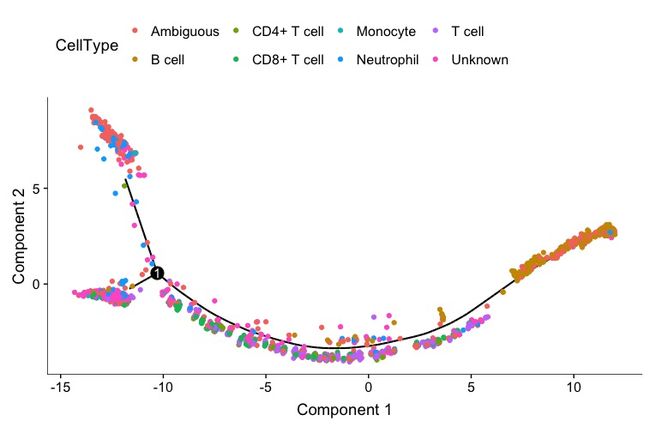
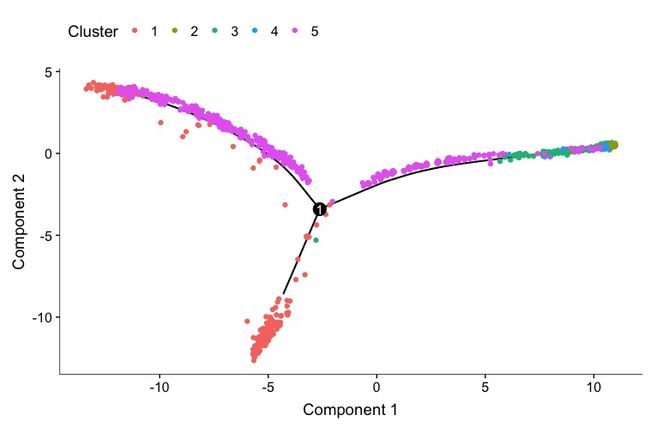
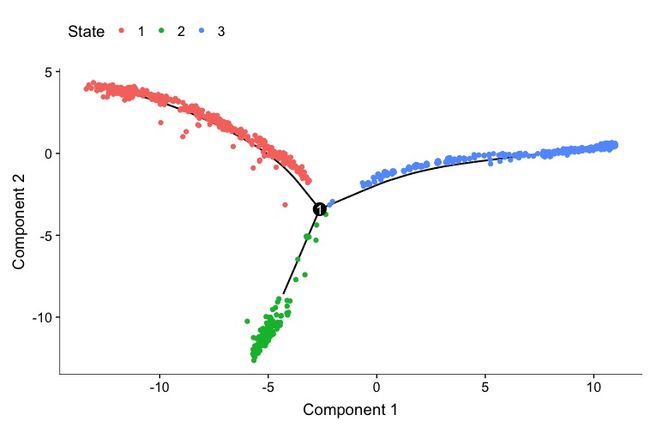
这是一种树状图,有三条细胞轨迹,表示细胞状态主要分为3个阶段,中间的数字1表示一个分叉。
上图的细胞依据不同的Cluster进行上色,根据之前的Seurat聚类分析,Cluster5(浅蓝色)对应的是中性粒细胞,在此图位于上面那一分支的顶端;Cluster0(红色)对应的是B细胞,主要位于右边一支的最顶端;左下角顶端的蓝色有可能是NK细胞,但不确定。这样看来比较适合做初始状态的是右边那一支。与下图对比得到的结果也差不多。
plot_cell_trajectory(clustered_spleen_monocle, color_by = "CellType") + facet_wrap(~CellType, nrow = 1)
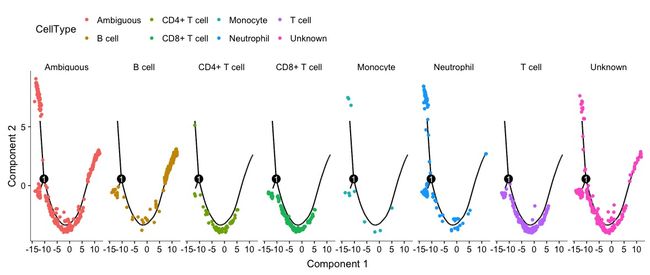
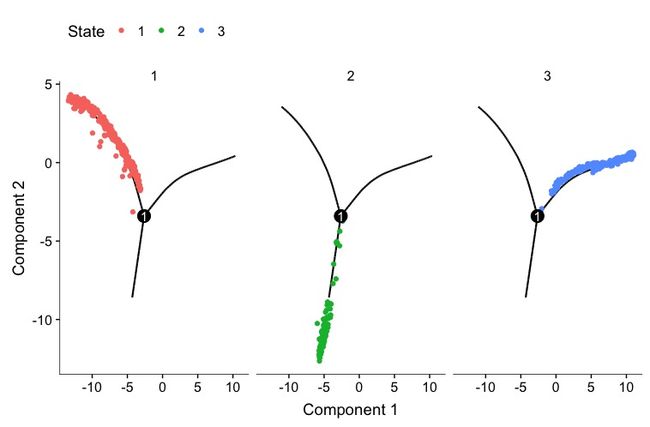
上图将每个细胞的分布轨迹分开显示,比较明显地看到B细胞集中的位置在右支顶端,然后集中为T细胞,中间掺杂一些中性粒细胞(也有可能没分好)。但是大部分的细胞还是没有分出来,这个结果还需要再处理一下。
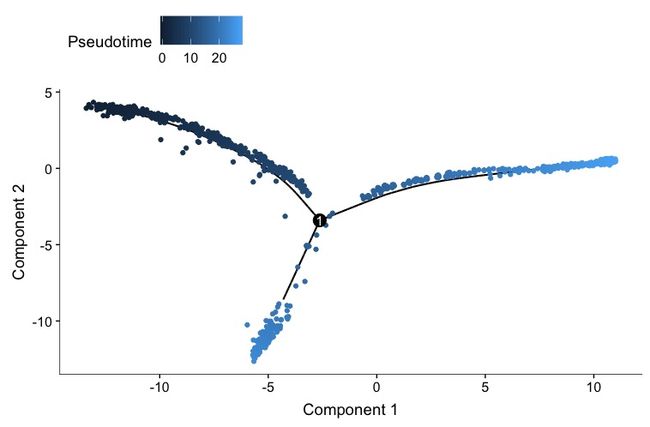
由于Monocle不能分辨哪一条轨迹才是“树根”,也就是不知道哪一个细胞状态才是更初始的,可设定root_state参数来设定哪条轨迹才是初始状态。然后赋予每一个细胞伪时间值,就可以观察基因在伪时间里的表达变化。待处理好细胞分类之后,就可以接着做这一步。
head(pData(clustered_spleen_monocle))
nGene nUMI orig.ident percent.mito Cluster Size_Factor CellType
AAACCTGAGGTGATAT 1072 3999 10X_spleen 0.01150863 3 0.8327676 T cell
AAACCTGAGTCATCCA 1562 4640 10X_spleen 0.03709295 5 0.9661104 Ambiguous
AAACCTGCAGTGAGTG 995 3489 10X_spleen 0.03955288 3 0.7269267 Monocyte
AAACGGGAGACTGGGT 1704 7397 10X_spleen 0.02852508 0 1.5411513 Ambiguous
AAACGGGAGACTTGAA 976 3102 10X_spleen 0.04932302 2 0.6462960 B cell
AAACGGGAGATAGGAG 1236 4548 10X_spleen 0.02682498 1 0.9475674 Ambiguous
情况②:未经过标准化处理的Seurat对象导入
创建CellDataSet对象(下简称CDS对象)
创建Seurat对象spleen_monocle,先去除一些测序质量差的细胞:
留下所有在>=3个细胞中表达的基因min.cells = 3;
留下所有检测到>=200个基因的细胞min.genes = 200。
spleen_monocle <- CreateSeuratObject(raw.data = spleen.data, min.cells = 3, min.genes = 200, project = "10X_spleen")
从Monocle中导入Seurat对象
library(monocle)
spleen_monocle <- importCDS(spleen_monocle, import_all = TRUE)
查看数据:
spleen_monocle
CellDataSet (storageMode: environment)
assayData: 15655 features, 1959 samples
element names: exprs
protocolData: none
phenoData
sampleNames: AAACCTGAGGTGATAT AAACCTGAGTCATCCA ... TTTGTCAGTCGTCTTC (1959 total)
varLabels: nGene nUMI orig.ident Size_Factor
varMetadata: labelDescription
featureData
featureNames: RP11-34P13.7 FO538757.2 ... AC240274.1 (15655 total)
fvarLabels: gene_short_name
fvarMetadata: labelDescription
experimentData: use 'experimentData(object)'
Annotation:
head(fData(spleen_monocle))
gene_short_name
RP11-34P13.7 RP11-34P13.7
FO538757.2 FO538757.2
AP006222.2 AP006222.2
RP4-669L17.10 RP4-669L17.10
RP11-206L10.9 RP11-206L10.9
LINC00115 LINC00115
head(pData(spleen_monocle))
nGene nUMI orig.ident Size_Factor
AAACCTGAGGTGATAT 1072 3999 10X_spleen NA
AAACCTGAGTCATCCA 1562 4640 10X_spleen NA
AAACCTGCAGTGAGTG 995 3489 10X_spleen NA
AAACGGGAGACTGGGT 1704 7397 10X_spleen NA
AAACGGGAGACTTGAA 976 3102 10X_spleen NA
AAACGGGAGATAGGAG 1236 4548 10X_spleen NA
15655个基因,1959个细胞,与之前创建的的Seurat对象相符。
计算SizeFactor和Dispersions
计算用于方便之后的分析使用。
spleen_monocle <- estimateSizeFactors(spleen_monocle)
spleen_monocle <- estimateDispersions(spleen_monocle)
数据清洗
根据前文所说的表达式,可以利用nUMI值进行过滤
head(pData(spleen_monocle))
nGene nUMI orig.ident Size_Factor num_genes_expressed
AAACCTGAGGTGATAT 1072 3999 10X_spleen 0.8207242 1070
AAACCTGAGTCATCCA 1562 4640 10X_spleen 0.9521386 1560
AAACCTGCAGTGAGTG 995 3489 10X_spleen 0.7164140 995
AAACGGGAGACTGGGT 1704 7397 10X_spleen 1.5188634 1704
AAACGGGAGACTTGAA 976 3102 10X_spleen 0.6369493 976
AAACGGGAGATAGGAG 1236 4548 10X_spleen 0.9338638 1236
观察到这里多了一个Size_Factor的列
#设置上下限:
upper_bound_spleen <- 10^(mean(log10(pData(spleen_monocle)$nUMI))
+ 2*sd(log10(pData(spleen_monocle)$nUMI)))
lower_bound_spleen <- 10^(mean(log10(pData(spleen_monocle)$nUMI))
- 2*sd(log10(pData(spleen_monocle)$nUMI)))
#可视化
qplot(nUMI,data = pData(spleen_monocle), geom = "density") + geom_vline(xintercept = lower_bound_spleen) + geom_vline(xintercept = upper_bound_spleen)
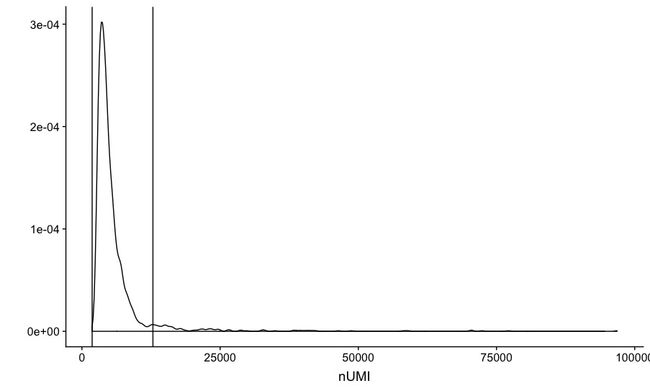
留下两条竖线中间的部分:
spleen_monocle <- spleen_monocle[,pData(spleen_monocle)$nUMI > lower_bound_spleen & pData(spleen_monocle)$nUMI < upper_bound_spleen]
spleen_monocle
CellDataSet (storageMode: environment)
assayData: 15655 features, 1864 samples
element names: exprs
protocolData: none
phenoData
sampleNames: AAACCTGAGGTGATAT AAACCTGAGTCATCCA ... TTTGTCAGTCGTCTTC (1864
total)
varLabels: nGene nUMI orig.ident Size_Factor
varMetadata: labelDescription
featureData
featureNames: RP11-34P13.7 FO538757.2 ... AC240274.1 (15655 total)
fvarLabels: gene_short_name
fvarMetadata: labelDescription
experimentData: use 'experimentData(object)'
Annotation:
过滤后剩下1864个细胞,15655个基因
数据标准化
# 将表达矩阵中所有值进行lognormalize.
L <- log(exprs(spleen_monocle[expressed_genes,]))
# Standardize each gene, so that they are all on the same scale,
# Then melt the data with plyr so we can plot it easily
library("reshape")
melted_dens_df <- melt(Matrix::t(scale(Matrix::t(L))))
# Plot the distribution of the standardized gene expression values.
qplot(value, geom = "density", data = melted_dens_df) +
stat_function(fun = dnorm, size = 0.5, color = 'red') +
xlab("Standardized log(FPKM)") +
ylab("Density")
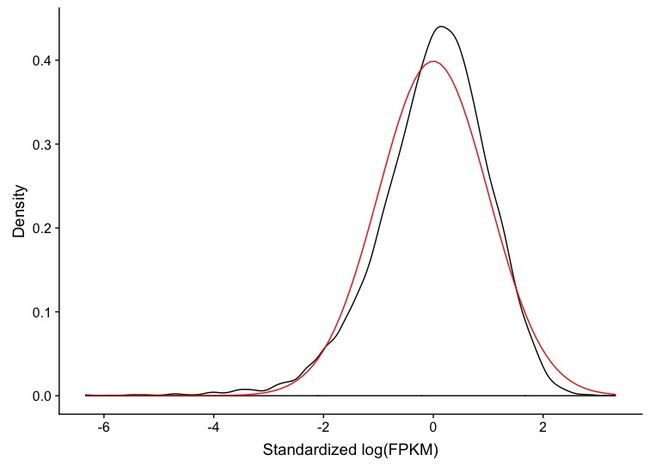
分类细胞
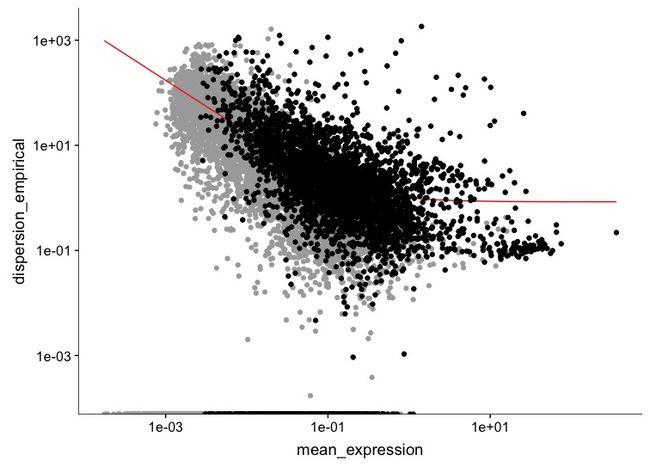
disp_table <- dispersionTable(spleen_monocle)
unsup_clustering_genes <- subset(disp_table, mean_expression >= 0.1)
spleen_monocle <- setOrderingFilter(spleen_monocle, unsup_clustering_genes$gene_id)
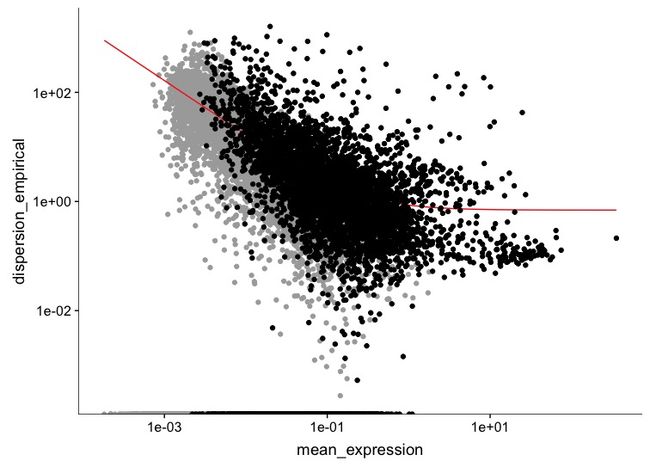
plot_ordering_genes(spleen_monocle)
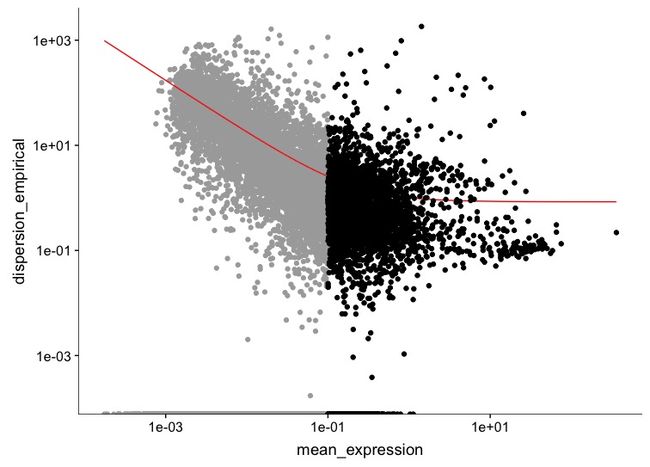
setOrderingFilter将一些用于之后聚类的基因标记起来;
plot_ordering_genes根据这些基因的平均表达水平展示了基因的表达差异程度,红线表示Monocle基于这种关系对分散的期望。我们用于聚类的基因显示为黑点,而其他基因显示为灰点。(这里有点不太明白纵坐标的dispersion经验值是什么意思)
聚类细胞
作碎石图:
plot_pc_variance_explained(spleen_monocle, return_all = F, max_components = 25)
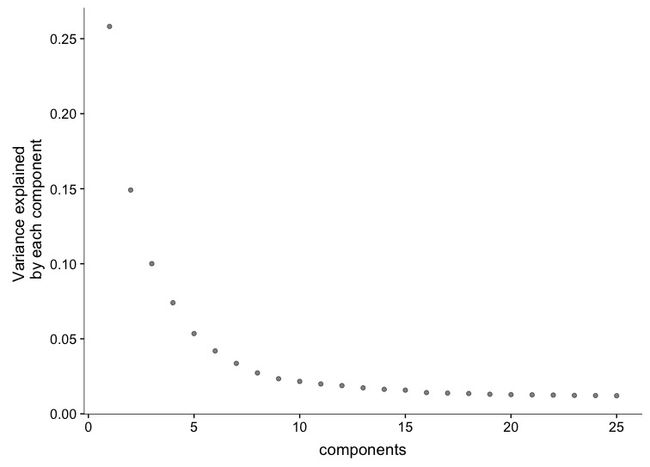
选择前8个成分进行聚类
spleen_monocle <- reduceDimension(spleen_monocle, max_components = 2, num_dim = 8, reduction_method = 'tSNE', verbose = T)
spleen_monocle <- clusterCells(spleen_monocle)
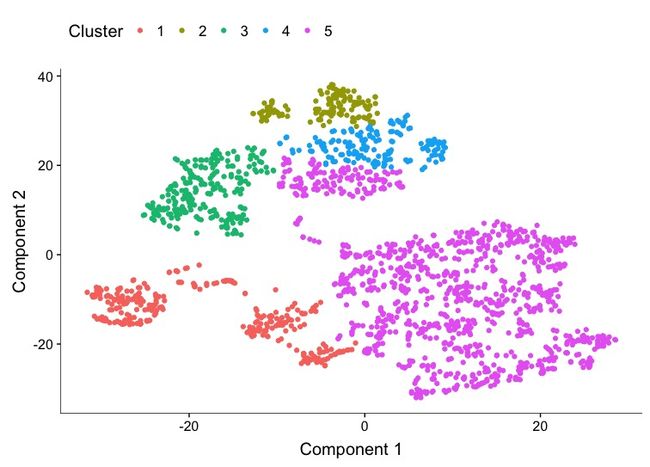
如果说每个时间点的细胞都聚在一起的话,那么大致上看此图正好分成4个模块。
将细胞按照伪时间的顺序排列在轨迹上
Step1:选择定义细胞发展的基因
这里可以用Monocle包中的dpFeature命令进行选择基因。
#这个命令运行时间特别长
diff_test_res <- differentialGeneTest(spleen_monocle,fullModelFormulaStr = "~Cluster")
ordering_genes <- row.names (subset(diff_test_res, qval < 0.01))
另一种方式是基于生物知识人为选择基因:
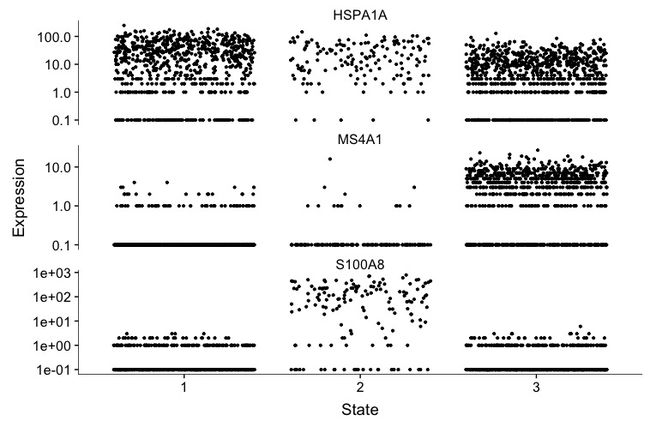
HSPA1A*基因是之前用Seurat包发现的一个比较有意思的基因,几乎在所有的cluster中都有不同程度的表达(下图),并且查阅文献发现此基因表达热休克蛋白,在机体发生缺血/缺氧时此蛋白的表达是一种保护机制,可以作为心脏骤停患者的预后标志物。还有学者研究基于脑缺血的持续时间跟HSPA1A表达量的关系,尽管该论文做了两组(缺血时间30分钟和60分钟)的最后的结论是这两组的表达量差异没有显著性,但是脾脏缺血是经过了12h, 24h 和72h的处理,时间跨度大很多,因此还是具有比较好的研究价值。
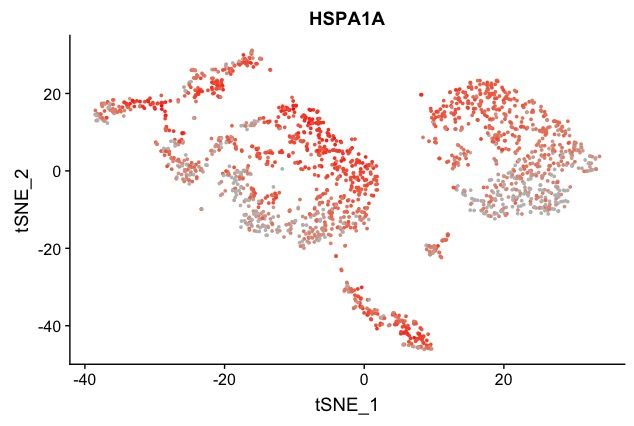
因此我认为选择HSPA1A作为反映细胞状态的标志基因也是可以的。
12月1号后——
Step2: 数据集降维
spleen_monocle <- setOrderingFilter(spleen_monocle, ordering_genes)
plot_ordering_genes(spleen_monocle)
spleen_monocle <- reduceDimension(spleen_monocle, max_components = 2, reduction_method = "DDRTree")
Step3: 将细胞按照伪时间排序
spleen_monocle <- orderCells(spleen_monocle)
colnames(pData(spleen_monocle))
[1] "nGene"
[2] "nUMI"
[3] "orig.ident"
[4] "Size_Factor"
[5] "Cluster"
[6] "peaks"
[7] "halo"
[8] "delta"
[9] "rho"
[10] "nearest_higher_density_neighbor"
plot_cell_trajectory(spleen_monocle, color_by = "Cluster")
plot_cell_trajectory(spleen_monocle, color_by = "State")
GM_state <- function(cds){
+ if (length(unique(pData(cds)$State)) > 1){
+ T0_counts <- table(pData(cds)$State, pData(cds)$Cluster)[,"0"]
+ return(as.numeric(names(T0_counts)[which
+ (T0_counts == max(T0_counts))]))
+ } else {
+ return (1)
+ }
+ }
plot_cell_trajectory(spleen_monocle, color_by = "Pseudotime")
plot_cell_trajectory(spleen_monocle, color_by = "State") +
+ facet_wrap(~State, nrow = 1)
作图
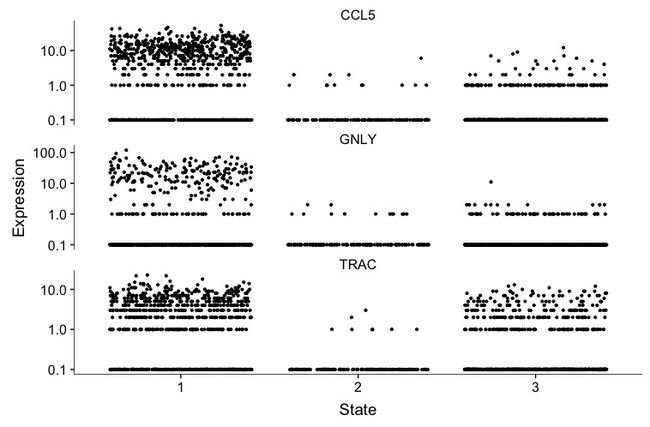
> blast_genes <- row.names(subset(fData(spleen_monocle), gene_short_name %in% c("CCL5","TRAC","GNLY")))
> plot_genes_jitter(spleen_monocle[blast_genes,],
+ grouping = "State",
+ min_expr = 0.1)
> blast_genes <- row.names(subset(fData(spleen_monocle), gene_short_name %in% c("HSPA1A","MS4A1","S100A8")))
> plot_genes_jitter(spleen_monocle[blast_genes,],
+ grouping = "State",
+ min_expr = 0.1)
下面是没有跑过的!!!
sp_expressed_genes <- row.names(subset(fData(spleen_monocle),
num_cells_expressed >= 10))
spleen_monocle_filtered <- spleen_monocle[HSMM_expressed_genes,]
my_genes <- row.names(subset(fData(spleen_monocle_filtered),
gene_short_name %in% c("CDK1", "MEF2C", "MYH3")))
cds_subset <- spleen_monocle_filtered[my_genes,]
plot_genes_in_pseudotime(cds_subset, color_by = "Cluster")
Reference
[1]Jenei, Z.M., Széplaki, G., Merkely, B. et al. Cell Stress and Chaperones (2013) 18: 447. https://doi.org/10.1007/s12192-012-0399-2
[2]Choi JI, Kim SD, Kim SH, Lim DJ, Ha SK. Semi-quantitative analyses of hippocampal heat shock protein-70 expression based on the duration of ischemia and the volume of cerebral infarction in mice. J Korean Neurosurg Soc. 2014;55(6):307-12.