今天推荐给大家一个R包WGCNA,针对我们的表达谱数据进行分析。
简单介绍:WGCNA首先假定基因网络服从无尺度分布,并定义基因共表达相关矩阵、基因网络形成的邻接函数,然后计算不同节点的相异系数,并据此构建系统聚类树。该聚类树的不同分支代表不同的基因模块(module),模块内基因共表达程度高,而分属不同模块的基因共表达程度低。
主要应用:如果某些基因在一个生理过程或不同组织中总是具有相类似的表达变化,那么我们有理由认为这些基因在功能上是相关的,可以把它们定义为一个模块(module)。当基因module被定义出来后,我们可以利用这些结果做很多进一步的工作,如筛选module的核心基因,关联性状,代谢通路建模,建立基因互作网络等。
好了,言归正传,我们开始一步步进行演示!
载入WGCNA包,设置随机种子,默认数据不进行因子转换
先把原始数据转列,转成横排是探针(基因),纵排是个体的顺序,先变成数列,用as.data.fame,然后改列名rownames(design) <- design[,1]
design <- design[,-1]
##datExpr<-as.data.frame(datExpr) (有可能需要先把数值转为数据集)
##> datExpr1<-read.table("test.txt",header=T,stringsAsFactors = F)
##> datExpr1<-t(datExpr1)
##> colnames(datExpr1)<-datExpr1[1,]
##> datExpr1<-datExpr1[-1,]
##> datExpr1<-as.data.frame(datExpr1)
library(WGCNA)
set.seed(1)
options(stringsAsFactors = F)
构造性状数据(亦或是分组数据)
obs<-paste("sam",1:10,sep="")
sample<-as.data.frame(diag(x=1,nrow = length(obs)))
rownames(sample)<-obs
colnames(sample)<-1:10
构造表达量数据
datExpr<-as.data.frame(t(matrix(runif(30000)+5,3000,10)))
rownames(datExpr)<-obs
names(datExpr)<-paste("transcript",1:3000,sep="")
明确样本数和基因数
nGenes = ncol(datExpr)
nSamples = nrow(datExpr)
首先针对样本做个系统聚类树
datExpr_tree<-hclust(dist(datExpr), method = "average")
par(mar = c(0,5,2,0))
plot(datExpr_tree, main = "Sample clustering", sub="", xlab="", cex.lab = 2,
cex.axis = 1, cex.main = 1,cex.lab=1)

针对前面构造的样品矩阵添加对应颜色
sample_colors <- numbers2colors(sample, colors = c("white","blue"),signed = FALSE)

构造10个样品的系统聚类树及性状热图
par(mar = c(1,4,3,1),cex=0.8)
plotDendroAndColors(datExpr_tree, sample_colors,
groupLabels = colnames(sample),
cex.dendroLabels = 0.8,
marAll = c(1, 4, 3, 1),
cex.rowText = 0.01,
main = "Sample dendrogram and trait heatmap")

这个有什么意义呢?
你可以将样本分为正常组和对照组,或者野生型和突变型等,从而可以查看样本聚类情况!
针对10个样品绘制主成分图(在这里不考虑分组情况)
pca = prcomp(datExpr)
sampletype<-rownames(sample)
par(mar = c(4,4,4,6))
plot(pca$x[,c(1,2)],pch=16,col=rep(rainbow(nSamples),each=1),cex=1.5,main = "PCA map")
text(pca$x[,c(1,2)],row.names(pca$x),col="black",pos=3,cex=1)
legend("right",legend=sampletype,ncol = 1,xpd=T,inset = -0.15,
pch=16,cex=1,col=rainbow(length(sampletype)),bty="n")

library(scatterplot3d)
par(mar = c(4,4,4,4))
scatterplot3d(pca$x[,1:3], highlight.3d=F, col.axis="black",color = rep(rainbow(nSamples),each=1),cex.symbols=1.5,cex.lab=1,cex.axis=1, col.grid="lightblue", main="PCA map", pch=16)
legend("topleft",legend = row.names(pca$x) ,pch=16,cex=1,col=rainbow(nSamples), ncol = 2,bty="n")

选择合适“软阀值(soft thresholding power)”beta
powers = c(1:30)
pow<-pickSoftThreshold(datExpr, powerVector = powers, verbose = 5)
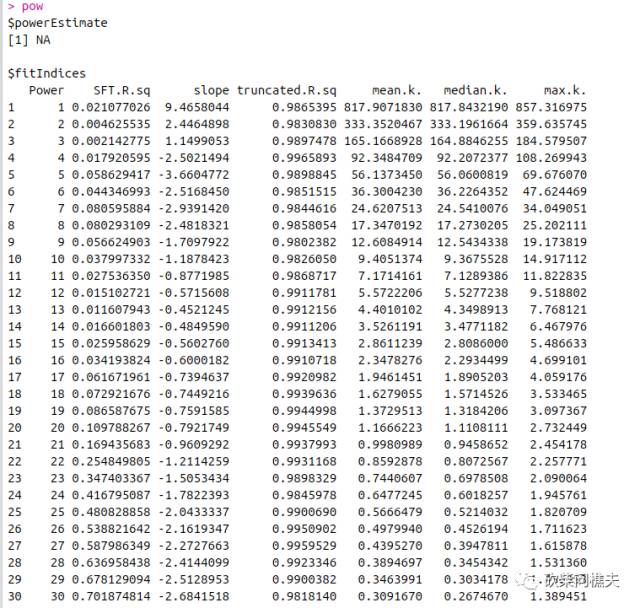
设置网络构建参数选择范围,计算无尺度分布拓扑矩阵
par(mfrow = c(1,2))
plot(pow$fitIndices[,1], -sign(pow$fitIndices[,3])*pow$fitIndices[,2],xlab="Soft Threshold (power)",ylab="Scale Free Topology Model Fit,signed R^2",type="n", main = paste("Scale independence"))
text(pow$fitIndices[,1], -sign(pow$fitIndices[,3])*pow$fitIndices[,2],labels=powers,cex=0.5,col="red")
plot(pow$fitIndices[,1], pow$fitIndices[,5],xlab="Soft Threshold (power)",ylab="Mean Connectivity", type="n",main = paste("Mean connectivity"))
text(pow$fitIndices[,1], pow$fitIndices[,5], labels=powers, cex=0.6,col="red")

参数beta取值默认是1:30,上述图形的横轴均代表权重参数β,左图纵轴代表对应的网络中log(k)与log(p(k))相关系数的平方。相关系数的平方越高,说明该网络越逼近无网路尺度的分布。右图的纵轴代表对应的基因模块中所有基因邻接函数的均值。
在这里,我们选择β=6构建基因网络。
接下来是非常重要的一块内容就是构架基因网络
大体思路:计算基因间的邻接性,根据邻接性计算基因间的相似性,然后推出基因间的相异性系数,并据此得到基因间的系统聚类树。然后按照混合动态剪切树的标准,设置每个基因模块最少的基因数目为30。根据动态剪切法确定基因模块后,再次分析,依次计算每个模块的特征向量值,然后对模块进行聚类分析,将距离较近的模块合并为新的模块。
1、计算树之间的邻接性
计算softPower
pow?
adjacency = adjacency(datExpr, power = softPower)
2、计算树之间的相异性
TOM = TOMsimilarity(adjacency)
dissTOM = 1-TOM
3、聚类分析
geneTree = hclust(as.dist(dissTOM), method = "average")
4、设置基因模块中最少基因包含30个
minModuleSize = 30
dynamicMods = cutreeDynamic(dendro = geneTree, distM = dissTOM, deepSplit = 2,pamRespectsDendro = FALSE, minClusterSize = minModuleSize)
5、基因分组上色
dynamicColors = labels2colors(dynamicMods)
table(dynamicColors)

6、计算基因模块的特征值
MEList = moduleEigengenes(datExpr, colors = dynamicColors)
MEs = MEList$eigengenes
MEDiss = 1-cor(MEs)
METree = hclust(as.dist(MEDiss), method = "average")
7、建立系统聚类树
MEDissThres = 0.4
plot(METree, main = "Clustering of module eigengenes", xlab = "", sub = "")
abline(h=MEDissThres, col = "red")

7、基因模块合并
merge = mergeCloseModules(datExpr, dynamicColors, cutHeight = MEDissThres, verbose = 3)
mergedColors = merge$colors
mergedMEs = merge$newMEs
8、绘制基因网络图
plotDendroAndColors(geneTree, cbind(dynamicColors, mergedColors), c("Dynamic Tree Cut", "Merged dynamic"), dendroLabels = FALSE, hang = 0.03, addGuide = TRUE, guideHang = 0.05)

拓展分析
不同模块基因热图及关键基因的表达
person=cor(datExpr,use = 'p')
corr<-TOM
Colors<-mergedColors
colnames(corr)<-colnames(datExpr)
rownames(corr)<-colnames(datExpr)
names(Colors)<-colnames(datExpr)
colnames(person)<-colnames(datExpr)
rownames(person)<-colnames(datExpr)
umc = unique(mergedColors)
lumc = length(umc)
for (i in c(1:lumc)){
if(umc[i]== "grey"){
next
}
ME=MEs[, paste("ME",umc[i], sep="")]
par(mfrow=c(2,1), mar=c(0.3, 5.5, 3, 2))
plotMat(t(scale(datExpr[,Colors==umc[i]])),nrgcols=30,rlabels=F,rcols=umc[i], main=umc[i], cex.main=2)
par(mar=c(5, 4.2, 0, 0.7))
barplot(ME, col=umc[i], main="", cex.main=2,ylab="eigengene expression",xlab="array sample")
}
一共会生成36个基因模块热图,由于篇幅有限,仅仅展示2个

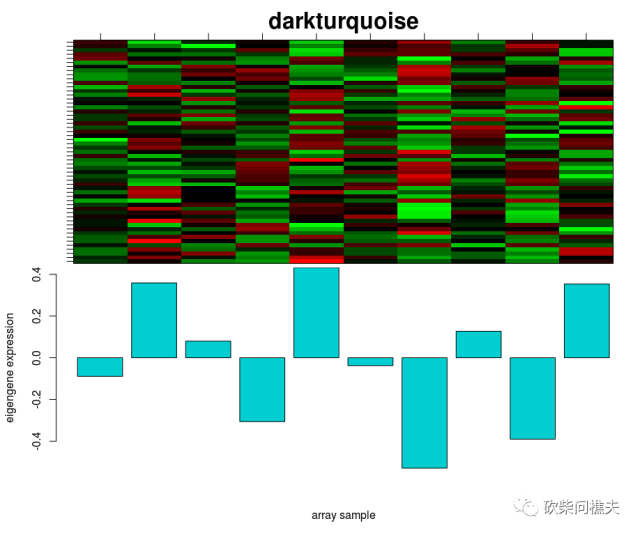
基因共表达网络热图
kME=signedKME(datExpr, mergedMEs, outputColumnName = "kME", corFnc = "cor", corOptions = "use = 'p'")
if (dim(datExpr)[2]>=1500) nSelect=1500 else nSelect=dim(datExpr)[2]
set.seed(1)
select = sample(nGenes, size = nSelect)
selectTOM = dissTOM[select, select]
selectTree = hclust(as.dist(selectTOM), method = "average")
selectColors = moduleColors[select]
plotDiss = selectTOM^7
TOMplot(plotDiss, selectTree, selectColors, main = "Network heatmap plot")
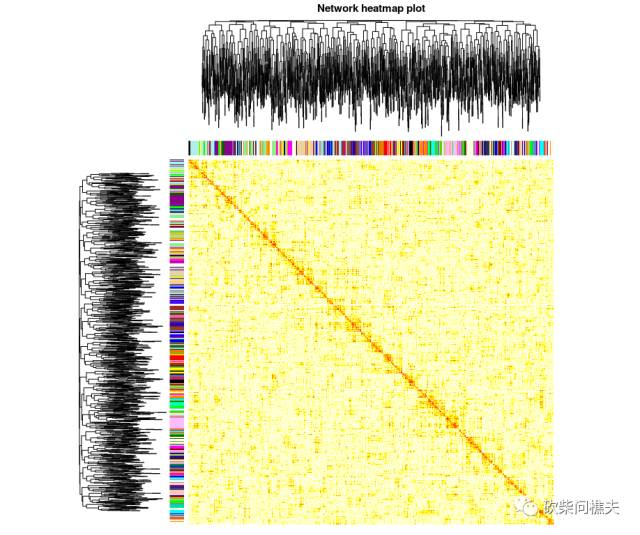
模块相关性热图
MEs = moduleEigengenes(datExpr, Colors)$eigengenes
MET = orderMEs(MEs)
plotEigengeneNetworks(MET, "Eigengene adjacency heatmap", marHeatmap = c(3,4,2,2), plotDendrograms = FALSE, xLabelsAngle = 90)

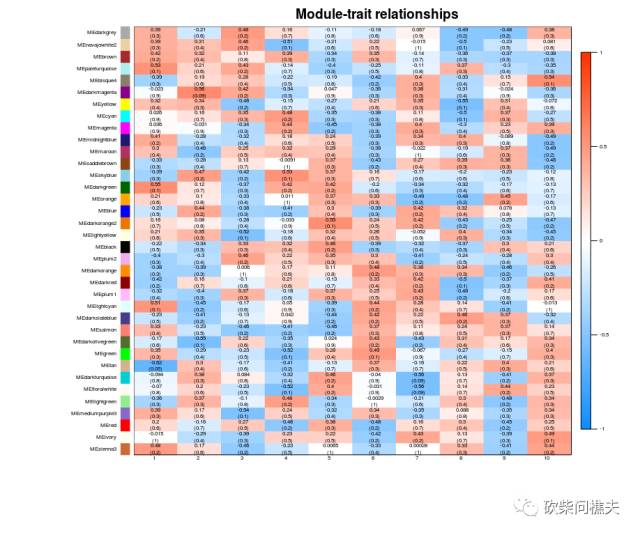
模块与性状相关性热图
moduleTraitCor = cor(MET, sample, use = "p")
moduleTraitPvalue = corPvalueStudent(moduleTraitCor, nSamples)
textMatrix = paste(signif(moduleTraitCor, 2), "\n(",signif(moduleTraitPvalue, 1), ")", sep = "")
dim(textMatrix) = dim(moduleTraitCor)
par(mar = c(2, 4, 2, 1.5))
labeledHeatmap(Matrix = moduleTraitCor,
xLabelsAngle = 0,
cex.lab = 0.5,
xLabels = colnames(sample),
yLabels = names(MET),
ySymbols = names(MET),
colorLabels = FALSE,
colors = blueWhiteRed(100),
textMatrix = textMatrix,
setStdMargins = FALSE,
cex.text = 0.5,
zlim = c(-1,1),
yColorWidth=0.02,
xColorWidth = 0.05,
main = paste("Module-trait relationships"))
基因的系统树图及性状相关性热图
geneTraitSignificance = as.data.frame(cor(datExpr, sample, use = "p"))
geneTraitColor=as.data.frame(numbers2colors(geneTraitSignificance,signed=TRUE,colors = colorRampPalette(c("blue","white","red"))(100)))
names(geneTraitColor)= colnames(sample)
par(mar = c(3.5, 7, 2, 1))
plotDendroAndColors(geneTree, cbind(mergedColors, geneTraitColor),
c("Module",colnames(sample)),dendroLabels = FALSE, hang = 0.03,
addGuide = TRUE, guideHang = 0.05)
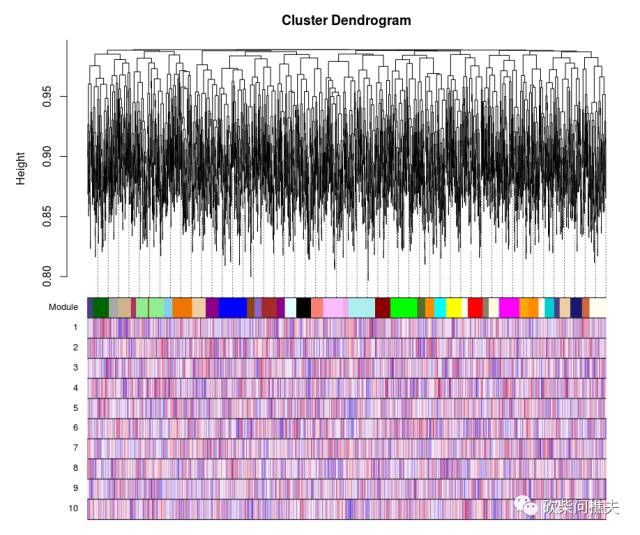
不同模块的基因显著性图
geneTraitSignificance = as.data.frame(cor(datExpr, sample, use = "p"))
GSPvalue = as.data.frame(corPvalueStudent(as.matrix(geneTraitSignificance), nSamples))
names(geneTraitSignificance) = paste("GS.", colnames(sample), sep="")
names(GSPvalue) = paste("GS.", colnames(sample), sep="")
modNames = substring(names(MET), 3)
for (module in modNames){
if(module== "grey"){ next }
column = match(module, modNames); # col number of interesting modules
moduleGenes = Colors==module;
par(mfrow = c(1,1))
verboseScatterplot(abs(kME[moduleGenes, column]),
abs(geneTraitSignificance[moduleGenes, 1]),
xlab = paste("Module Membership in", module, "module"),
ylab = "Gene significance",
main = paste("Module membership vs. gene significance
"),cex.main = 1.2, cex.lab = 1.2, pch=19,cex.axis = 1.2, col = module)}

好了,今天就到这里……