R包EBSeq差异基因分析
R包EBSeq差异基因分析
上个星期师妹问我有没有一种R包,可以根据相对丰度筛选差异基因(最常用的筛选差异基因的R包如edgeR、DESeq2等,只能识别整数数值的基因丰度信息,无法基于小数类型的基因相对丰度信息计算差异)。然后我试着找了一下,看到有说EBSeq包可以使用非整数的reads counts值进行差异基因的计算,于是就测试了下。
EBSeq包中的方法确实能够将非整数的基因丰度数据作为输入文件,但是当小数位数过多,还有低于1的小数过多时貌似仍然不可行(会提示错误),也就是说这个包仍然不能根据相对丰度筛选差异基因。尽管它可以识别非整数值,感觉应该是指的在整数类型的基础上经过某些标准化后的基因丰度数据(标准化后产生了小数),与相对丰度还是不一样的。
尽管未能如愿,但是意外地发现了EBSeq居然可以比较多组间的差异,没错,它不局限于只能在两组间筛选差异基因。而且,它还提供了图形化操作界面的插件供我们选择,这样也方便了不熟练R操作的同学们可以通过点击按钮的方式完成统计分析。
总之EBSeq是个非常不错的R包,下面就对EBSeq进行差异基因分析的方法作个简介吧。个人表达习惯,下文统一使用“基因丰度”这个名词。
EBSeq应用经验贝叶斯分层模型(empirical Bayes hierarchical model),寻找组间差异表达的gene(基因水平)或isoforms(可理解为由同一个基因编码的不同蛋白,由可变剪切产生)。
EBSeq网站链接:https://www.biostat.wisc.edu/~kendzior/EBSEQ/
安装方法、帮助文档、插件信息、参考文献、更新日志等均可在此界面中找到。
其中,帮助文档链接:http://www.bioconductor.org/packages/devel/bioc/vignettes/EBSeq/inst/doc/EBSeq_Vignette.pdf
本文仅对EBSeq做差异基因筛选的一般流程作简要说明。关于EBSeq所涉及的统计模型算法,该R包更多功能,以及常见问题及注意事项等,可参阅帮助文档详细了解。
本文中的R代码已上传至百度盘,提取码xvfh:https://pan.baidu.com/s/1-DbQkWIkiCyEn2o9H4xDfA
安装及加载EBSeq包
见其网站中的描述,EBSeq可直接使用Bioconductor安装。Bioconductor安装过程一般没啥大问题,要是有些依赖的包没能自动安装成功,大不了手动安装即可。
#Bioconductor 安装 EBSeq
source('http://bioconductor.org/biocLite.R')
biocLite('EBSeq')
#如果连接失败可更改为 source('https://bioconductor.org/biocLite.R')
#加载
library(EBSeq)
后面有可能在加载EBSeq时会遇到这么一个问题,blockmodeling包载入时提示如下错误。

根据提示,我们在相应的路径下找到“CITATION”这个文件,然后用记事本等工具打开它,即可知道报错的原因:里面出现了R环境无法识别的字符(作者名称)。解决起来也很方便,把这些字符删掉就行了(删掉、或者替换成其它可以被R识别的字符都可以,而使用“#”注释掉是没用的)。不过由于无法被R环境识别的字符不止出现了一行,因此我们就简单粗暴点吧,把三个“citEntry()”里面的字符全删光得了(虽然觉得有些对不起作者……),肯定就没问题了。

EBSeq差异基因分析
EBSeq包成功加载后,我们以EBSeq包中自带的示例数据为例,简要展示差异基因分析流程。以下分别以两组间差异基因分析以及三组间差异基因分析来演示(两组模式和多组模式的方法有些区别,详见下文)。
两组间差异基因分析
基因数据集及样本分组信息
测试数据集GeneMat,是一个模拟的基因丰度数据矩阵,包含10列样本共计1000行基因。我们载入该数据集,并手动将该数据集中的样本划分为两组,前5列的样本定义为C1组,后5列的样本定义为C2组,以期使用EBSeq的方法计算C1组与C2组中的差异基因作为演示。
#测试数据
data(GeneMat)
#手动将测试基因数据集设置为两组,前 5 列为 C1 组,后 5 列 C2 组
group <- rep(c('C1', 'C2'), each = 5)

标准化因子
在差异基因分析前,需要分别对每个样本指定一个标准化因子(sample-specific normalization factor,在EBSeq中称为library size factor,简写为ls),用于对数据标准化。详细信息请参阅DESeq帮助文档。标准化方法根据实际情况来确定,常用的标准化方法例如中位数标准化(Median Normalization)、分位数标准化(Quantile Normalization)、缩放标准化(Scaling Normalization)等。
此处对于示例数据基因丰度数据矩阵GeneMat,选择使用了中位数标准化方法。
#通过 MedianNorm() 来获取标准化因子,等同于 DESeq 中的中位数标准化(Median Normalization)方法 ls_factor <- MedianNorm(GeneMat) #其它标准化方法,例如通过 QuantileNorm() 实现上四分位数标准化 #ls_factor <- QuantileNorm(GeneMat, 0.75) #等等方法,根据实际情况选择,大家自行了解
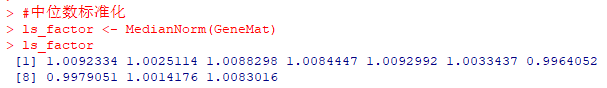
结果返回一组向量。此处共包含10个元素(中位数标准化因子),分别对应了10个样本。
EBSeq差异基因分析
使用EBTest()函数运行两组间的EBSeq差异分析流程。
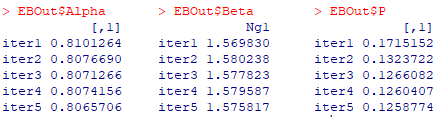
#EBSeq 差异基因分析,本示例迭代 5 次是合适的 EBOut <- EBTest(Data = GeneMat, Conditions = factor(group), sizeFactors = ls_factor, maxround = 5)
Data,指定基因丰度数据矩阵GeneMat;Conditions,指定样本分组信息group,最好提前转化为因子类型(虽然直接输入一组字符向量也可以,它也会自动转为因子类型,但是分组因子的前后顺序就无法自己确定了),并确保group中的分组名称顺序与GeneMat中的样本名称顺序(列顺序)对应;sizeFactors,输入上述所得的样本标准化因子,以实现对数据的标准化;maxround = 5,对于本示例迭代5次计算,真实的数据分析中还需根据实际情况选择迭代次数(迭代次数的选择是否合理见下文)。其它参数我们使用了默认参数,更多详情可使用?EBTest查看R文档。
备注:对于示例的分组信息字符向量group,包含分组C1和C2。这里我们在使用as.factor()将它转化为因子类型后,默认顺序C1在前C2在后,因此会计算C1中的基因相对于C2上调了还是下降了,这点需要注意,实际的数据分析中不要弄反了。因此,这里建议提前将group转化为因子类型,并在转化时确定好分组因子的前后顺序(factor()中使用levels参数指定)。

可通过summary(EBOut),查看计算结果中都包含哪些信息,有兴趣可自行研究结果的各部分都表示了什么内容。例如,C1Mean和C2Mean分别为两个分组中,各基因标准化后的平均丰度。

可通过这3个指标判断选择的迭代次数maxround是否合理,一般来说最后两个值相差小于0.01就可以了。
提取差异基因
通过GetDEResults()函数,获取差异基因。
#鉴定差异基因(FDR < 0.05,其他参数如 Fold Change 等使用默认值) EBDERes <- GetDEResults(EBOut, FDR = 0.05)
在该函数中,我们指定参数FDR = 0.05,可以理解为在执行FDR校正后以p<0.05为标准鉴定差异基因。其它参数使用默认值其它参数我们在这里使用默认值(例如Fold Change阈值等)。对于该函数的更多细节,请使用?GetDEResults参阅R文档。
结果赋值给列表EBDERes,包含3项内容。DEfound为鉴定的差异基因,对于示例数据我们共鉴定到了95个差异基因;PPMat包含两列信息,PPEE(within-condition variability的后验概率)和PPDE(across-condition variability的后验概率),如果PPEE<0.05(即可以理解为PPDE显著高于PPEE),即可将该基因定义为差异基因(DE gene);Status则记录了每种基因的判别状态,包含DE(差异)、EE(无差异)、Filtered: Low Expression、Filtered: Fold Change、Filtered: Fold Change Ratio(后面三种表示该基因被过滤掉)五种类型。
当然,GetDEResults()肯定不是简单地只根据FDR筛选的,还会考虑Fold Change (FC)值等信息(在这里我没设置其他参数,均使用默认值),具体详见?GetDEResults帮助。

既然根据PPEE<0.05的标准判别差异基因,那么即可知这里的PPEE其实就相当于FDR p-value了。也就是说上述方法中,我们默认根据FDR p-value < 0.05判断是否为差异基因。当然,若你想提高筛选标准,如FDR p-value < 0.01水平,更改参数为“GetDEResults(EBOut, FDR = 0.01)”即可。
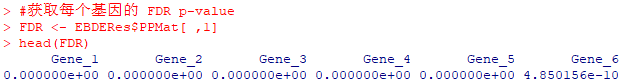
#提取每个基因的 FDR p-value FDR <- EBDERes$PPMat[ ,1]
提取Fold Change (FC)值
对于Fold Change (FC)值,可以通过PostFC()函数获得。对于该函数的更多信息,可使用?PostFC参阅R文档。
#获取获取每个基因的 Fold Change (FC) gene_FC <- PostFC(EBOut)
结果中包含RealFC和PostFC。二者的区别在于,RealFC基于原始数据获得,PostFC基于标准化后的数据获得,因此PostFC(posterior fold)会给低丰度的基因提供一个较低的极值,以降低低丰度基因的重要性。可以详见帮助文档。例如,如果gene1的mean1=5000且mean2=1000,则其RealFC和PostFC相差不大,大致均为5;如果gene2的mean1=5且mean2=1,则其RealFC为5,但PostFC将远低于5而接近1;此时当我们按PostFC进行排序时,gene2将不如gene1重要。根据实际需要,选择使用RealFC或PostFC。

可以通过PlotPostVsRawFC()比较每个基因的RealFC和PostFC。
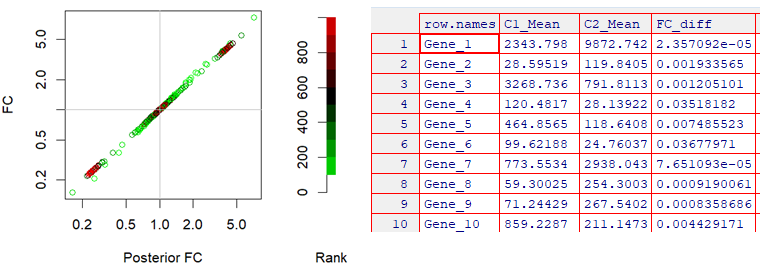
#Plot PostFC vs RealFC PlotPostVsRawFC(EBOut, gene_FC) #不妨直接观察基因丰度水平与 |RealFC - PostFC| 值的关系 FC_diff <- data.frame(C1_Mean = unlist(EBOut$C1Mean), C2_Mean = unlist(EBOut$C2Mean), FC_diff = abs(gene_FC$RealFC-gene_FC$PostFC))
下面的点图展示了示例数据中1000个基因的RealFC(FC)和PostFC(Posterior FC)的关系,基因的根据cross-condition mean(adjusted by the normalization factors)排序(升序排列,因此低丰度的基因排在前面,展示为绿色;高丰度基因排在后面,展示为红色)。越低丰度的基因,RealFC和PostFC差别越明显,因此越偏离斜对角线。这就很明了地为我们展示了PostFC在缩小低丰度基因差异方面的重要性。
或者,我们直接统计RealFC和PostFC的差值的绝对值,并结合各基因在C1组和C2组中的丰度一起展示。在结果中可以轻易地看出,越低丰度的基因,RealFC和PostFC的差值的绝对值越大。
模型诊断
EBSeq依赖于参数假设,因此在每次分析时需要执行检查。可使用Q-Q图,拟合经验q's和使用Beta先验分布模拟的q's,评估参数假设Beta先验的合理性,如果两个条件下的数据点位于y = x线上,则表明Beta先验是合理的。或者,根据密度图来判断,如果二者趋势一致,则表明Beta先验合理。
#使用 Q-Q 图,拟合经验 q's 和使用 β 先验分布模拟的 q's,评估 β 先验合理性 par(mfrow = c(1, 2)) QQP(EBOut) #查看经验 q's 和使用 β 先验分布模拟的 q's 的密度图,评估 β 先验合理性 par(mfrow = c(1, 2)) DenNHist(EBOut)


整合输出结果
确认以上结果没什么问题的话,不妨对以上的主要统计结果做个整合,输出在本地,便于浏览结果数据并在后续加以筛选等。
#整合版输出示例 C1_Mean <- unlist(EBOut$C1Mean) #C1 分组中,各基因标准化后的平均丰度 C2_Mean <- unlist(EBOut$C2Mean) #C2 分组中,各基因标准化后的平均丰度 FoldChange <- gene_FC$PostFC #使用 PostFC 获取 log2FoldChange <- log(FoldChange, 2) #log2 Fold Change EBSeq_stat <- data.frame(C1_Mean, C2_Mean, FoldChange, log2FoldChange, FDR) write.csv(EBSeq_stat, 'EBSeq_stat_two.csv', quote = FALSE)
这里我们保存为csv格式,“EBSeq_stat_two.csv”的内容如下所示。第一列为各基因的id;C1_Mean和C2_Mean列为两个分组中,各基因标准化后的平均丰度;FoldChange列就是Fold Change (FC)值了,我们使用基于标准化后的数据获得的FC值,即PostFC;log2FoldChange就是对FoldChange作了log2转化;FDR,也就是上文中提到的PPEE值,这里相当于FDR p-value。如果你想输出更多的信息,自定义编写代码另行添加所需的内容。

至于后面怎样对数据进行下一步的处理,大家随便了。比方说,上文中通过EBSeq的“GetDEResults(data, FDR = 0.05)”默认方法鉴定到了95个差异基因,我们可以直接使用;或者再更改GetDEResults()的参数设置,适当调整FDR或者FoldChange阈值等。如果想先将EBSeq的计算结果导出,然后再根据FoldChange和FDR进行手动筛选也可以,例如依据FoldChange≥2以及FDR<0.01水平在导出的表格中自定义筛选;或者只关注FDR,不考虑FoldChange;等等方法自己视情况确定标准。
差异火山图展示EBSeq分析结果
如果你是根据FoldChange和FDR筛选的目标基因,那么可以选择使用火山图来进行可视化展示。这也是很多文献中经常使用的图。
本人习惯使用ggplot2,差异火山图在ggplot2中其实就是散点图类型了。一个作图示例如下,依据FoldChange≥2以及FDR<0.05的标准作图展示差异基因类型。
library(ggplot2)
#根据 FC >= 2 和 FDR < 0.05 标记差异分析类型,赋值颜色
EBSeq_stat[which(EBSeq_stat$FDR < 0.05 & abs(EBSeq_stat$log2FoldChange) >= 0.5),'color2'] <- 'blue2'
EBSeq_stat[which(EBSeq_stat$FDR < 0.05 & abs(EBSeq_stat$log2FoldChange) < 0.5),'color2'] <- 'green3'
EBSeq_stat[which(EBSeq_stat$FDR >= 0.05 & abs(EBSeq_stat$log2FoldChange) >= 0.5),'color2'] <- 'red2'
EBSeq_stat[which(EBSeq_stat$FDR >= 0.05 & abs(EBSeq_stat$log2FoldChange) < 0.5),'color2'] <- 'gray30'
#ggplot2 作图示例
volcano_plot <- ggplot(EBSeq_stat, aes(log2FoldChange, -log(FDR + min(FDR[FDR != 0], na.rm = TRUE), 10))) + #给 FDR 加个小值,避免 0 无法 log 转化的问题
geom_point(aes(color = color2), alpha = 0.6, show.legend = FALSE) +
scale_colour_manual(values = EBSeq_stat$color2, limits = EBSeq_stat$color2) +
theme(panel.grid = element_blank(), panel.background = element_rect(color = 'black', fill = 'transparent')) +
geom_vline(xintercept = c(-0.5, 0.5), color = 'gray', size = 0.5) +
geom_hline(yintercept = -log(0.05, 10), color = 'gray', size = 0.5) +
labs(x = 'log2 Fold Change', y = '-log10 FDR')
#ggsave('volcano_plot.pdf', volcano_plot, width = 5, height = 5)
ggsave('volcano_plot.png', volcano_plot, width = 5, height = 5)
首先根据FoldChange和FDR的值,标记各基因的差异类型,以及赋值对应的颜色等,然后加载ggplot2绘制散点图,横轴展示log2 Fold Change,纵轴展示-log10 FDR。作图时不要忘记对0值做下处理,避免无法被log转化的问题。作图结果如下所示。

其它可视化方案
其它的可视化方案还可选:柱形图、箱线图、热图等,视情况加以考虑吧。
例如,以下使用热图展示前30个差异基因在10个样本中的丰度信息。
##前 30 个差异基因的热图展示示例
#选择前 30 个差异基因
DEgene_30_name <- head(EBDERes$DEfound, 30)
DEgene_30_count <- GeneMat[DEgene_30_name, ] #原始丰度
names(DEgene_30_count) <- as.character(1:10)
colgroup <- rep(c('red', 'blue'), each = 5)
#gplots 包 headmap.2()
library(gplots)
colorsChoice <- colorRampPalette(c('red', 'white', 'blue'))
heatmap.2(DEgene_30_count, density.info = c('none'), trace = 'none',col = colorsChoice(20), scale = 'column', srtCol = 0, ColSideColors = colgroup, colCol = colgroup) #作图时默认对基因丰度标准化

多组间差异基因分析(以三组间比较为例)
个人感觉这是这个R包做的最好的地方,提供了可以进行多组间比较的方法。这样,我们就用不着再单独执行两两分组间的比较并最后再整合了(回想当初使用过DESeq2,仅能在两组间筛选差异基因,想扩展到三组间时很麻烦,就想着有没有其他方法,然后找了N久没能找到,居然在这里偶然间发现了)。
和上面的示例一样,这里同样使用EBSeq自带的数据集进行演示。
数据准备(同上文,基因集、分组信息、标准化因子等)
测试数据集MultiGeneMat,是一个模拟的基因丰度数据矩阵,包含6列样本共计500行基因。我们载入该数据集,并手动将该数据集中的样本划分为三组,前2列的样本定义为C1组,中间2列为C2组,最后2列为C3组。同时获取标准化因子(标准化因子的描述见上文)。
#测试数据
data(MultiGeneMat)
#分组,模拟 3 组
group <- rep(c('C1', 'C2', 'C3'), each = 2)
#通过 MedianNorm() 来获取标准化因子,等同于 DESeq 中的中位数标准化(Median Normalization)方法
ls_factor <- MedianNorm(MultiGeneMat)

预指定差异模式(类型)
多组间比较的方法与两组间比较有些区别,需要预先指定基因差异模式。后面在计算时,对计算结果作判断,根据预先指定的基因差异模式划分归类。比方说本示例中共设置了3个分组(C1、C2、C3),那么基因差异模式就有5种。
#基因差异模式 Pattern parti <- GetPatterns(group) PlotPattern(parti)

Pattern1,某基因在3组间无差异;Pattern2,某基因在C3组与其它两组显著不同(无论是它高了还是低了,都算);Pattern3,某基因在C2组与其它两组显著不同(无论是它高了还是低了,都算);Pattern4,某基因在C1组与其它两组显著不同(无论是它高了还是低了,都算);Pattern5,某基因在C1、C2、C3组之间均存在显著不同(无论该基因在各分组中的丰度C1>C2>C3、C1>C3>C2、C3>C1>C2等等6种组合顺序,都算)。这个还是不难理解的吧。
好了,在实际的分析中,我们可能不关注这么多情况,那么可以在所有可能的差异模式中做下筛选。此外我们还很容易获知,当分组很多时,可能出现的差异模式(组合)也会很多,全部用于计算也会相当消耗资源,那么也不妨过滤一些不必要的组合。比方说这里我们只想关注Pattern1和Pattern5这两类的话,可以这样筛选下。

EBSeq差异基因分析
在大于两组的情况下,使用EBMultiTest()函数运行EBSeq差异分析流程。它的用法和上文中的EBTest()(两组情形)大致一致,详情使用?EBMultiTest参阅R文档了解。
#EBSeq 计算,本示例迭代 5 次是合适的 MultiOut <- EBMultiTest(Data = MultiGeneMat, Conditions = factor(group), sizeFactors = ls_factor, AllParti = parti, maxround = 5)
Data,指定基因丰度数据矩阵MultiGeneMat;Conditions,指定样本分组信息group,并转化为因子类型(这时其实你可以直接输入一组字符向量,它也会自动转为因子类型,分组因子的顺序在多组比较时不重要了,因为在这种模式下只观察“是否有差异”,而不具体关注“谁相对谁上升/下降”),并确保group中的分组名称顺序与MultiGeneMat中的样本名称顺序(列顺序)对应;sizeFactors,输入上述所得的样本标准化因子,以实现对数据的标准化;AllParti,指定基因差异模式,在本示例中为了更详细演示过程,使用了所有可能的差异模式(未作筛选,由于示例数据不是很大,分组也不多,因此运行起来也不慢);maxround = 5,对于本示例迭代5次计算,真实的数据分析中还需根据实际情况选择迭代次数(迭代次数的选择是否合理,在上文的方法中已有提到)。其它参数我们使用了默认参数。
可通过summary(MultiOut),查看计算结果中都包含哪些信息,有兴趣可自行研究结果的各部分都表示了什么内容,不再多说。

提取差异基因
在多组比较模式下,使用GetMultiPP()函数来获得每个基因在每种可能的差异模式中的后验概率。细节部分使用?GetMultiPP查看说明。
#获得每个基因在每个 Pattern 中的后验概率 MultiPP <- GetMultiPP(MultiOut)
结果赋值给列表MultiPP,包含3项内容。Patterns为先前预指定的基因差异模式;PP为各基因在各模式中的后验概率;MAP为最终划分的各基因的Pattern,根据PP作判断,一般来讲哪个Pattern的后验概率高就划分为哪种模式了。

关于多组比较中的Fold Change (FC)
多组比较模式下,无法再通过PostFC()获取Fold Change值(上文两组间BESeq分析中使用它获取)。需要的话,我们可以手动计算。在上一步,我们执行了多组间的EBSeq差异分析后将结果赋值给MultiOut。因此,我们可以从中提取出每个分组中各基因标准化后的平均丰度(存储在MultiOut$SPMean中),并据此计算两两分组间的Fold Change值。
不过貌似这样也挺麻烦的,而且多组比较中的计算方法也与两组间比较有些差别,不便于再根据Fold Change值等继续去筛,因此也就不再多说了。
模型诊断
可参考上文方法。
整合输出结果
我们对以上的主要统计结果做个整合,输出在本地,便于浏览结果数据并在后续加以筛选等。
备注:在多组比较模式下,各分组中各基因标准化后的平均丰度数值,会存储在同一个list中。对于上述示例,就是list“MultiOut$SPMean”,其中又包含了3个list [1]、[2]、[3]。由于在一开始我们指定的样本分组信息(字符向量)group在转化为因子类型后,默认的分组因子顺序为C1、C2、C3,因此“MultiOut$SPMean”中的子list [1]、[2]、[3]即分别对应分组C1、C2、C3。实际的数据分析中注意这点,不要提取错数据。
#整合并输出,一个示例 C1_Mean <- unlist(MultiOut$SPMean[[1]][[1]]) #C1 分组中,各基因标准化后的平均丰度 C2_Mean <- unlist(MultiOut$SPMean[[1]][[2]]) #C2 分组中,各基因标准化后的平均丰度 C3_Mean <- unlist(MultiOut$SPMean[[1]][[3]]) #C3 分组中,各基因标准化后的平均丰度 MAP <- MultiPP$MAP #Pattern 类型 EBSeq_stat <- data.frame(C1_Mean, C2_Mean, C3_Mean, MAP) for (i in 1:nrow(EBSeq_stat)) EBSeq_stat[i,'C1_C2_C3'] <- paste(parti[EBSeq_stat[i,'MAP'], ], collapse = '_') write.csv(EBSeq_stat, 'EBSeq_stat_three.csv', quote = FALSE)
这里我们保存为csv格式,“EBSeq_stat_three.csv”的内容如下所示。第一列为各基因的id;C1_Mean、C2_Mean、C3_Mean列为3个分组中,各基因标准化后的平均丰度;MAP和C1_C2_C3为Pattern类型。当然,如果你想输出更多的信息,自定义编写代码另行添加所需的内容。

同样地,至于后面怎样对数据进行下一步的处理,大家随便了。一般来讲,多组比较中考虑的东西越多,分析起来也就越繁琐。我本人还是比较推荐直接使用它给出的Pattern结果作判别了。
三元相图展示EBSeq分析结果
三组间比较结果的可视化,可以考虑使用三元相图呈现,效果挺不错的。三元相图的基本描述及绘图方法可参考http://blog.sciencenet.cn/blog-3406804-1173937.html。
以下为一个三元相图的作图示例,使用ggtern绘制(ggplot2的衍生包,语法和ggplot2几乎一致,如果你也喜欢ggplot2的风格,就可以考虑它)。
##三个样本的可视化可以考虑使用三元相图
#这里不再关注 Fold Change (FC),直接根据 Pattern,以及各基因在各分组中的平均丰度,判别基因富集情况
#计算各基因在哪个分组中的丰度最高
EBSeq_stat$max <- apply(EBSeq_stat[1:3], 1, function(x) names(x)[x %in% max(x)])
#根据 Pattern 以及各基因所在丰度最高的分组,标记基因富集分组
EBSeq_stat[which(EBSeq_stat$MAP == 'Pattern1'),'high_sample'] <- 'none'
EBSeq_stat[which(EBSeq_stat$MAP == 'Pattern2' & EBSeq_stat$max == 'C3_Mean'),'high_sample'] <- 'C3'
EBSeq_stat[which(EBSeq_stat$MAP == 'Pattern2' & EBSeq_stat$max %in% c('C1_Mean', 'C2_Mean')),'high_sample'] <- 'C1 & C2'
EBSeq_stat[which(EBSeq_stat$MAP == 'Pattern3' & EBSeq_stat$max == 'C2_Mean'),'high_sample'] <- 'C2'
EBSeq_stat[which(EBSeq_stat$MAP == 'Pattern3' & EBSeq_stat$max %in% c('C1_Mean', 'C3_Mean')),'high_sample'] <- 'C1 & C3'
EBSeq_stat[which(EBSeq_stat$MAP == 'Pattern4' & EBSeq_stat$max == 'C1_Mean'),'high_sample'] <- 'C1'
EBSeq_stat[which(EBSeq_stat$MAP == 'Pattern4' & EBSeq_stat$max %in% c('C2_Mean', 'C3_Mean')),'high_sample'] <- 'C2 & C3'
EBSeq_stat[which(EBSeq_stat$MAP == 'Pattern5' & EBSeq_stat$max == 'C1_Mean'),'high_sample'] <- 'C1'
EBSeq_stat[which(EBSeq_stat$MAP == 'Pattern5' & EBSeq_stat$max == 'C2_Mean'),'high_sample'] <- 'C2'
EBSeq_stat[which(EBSeq_stat$MAP == 'Pattern5' & EBSeq_stat$max == 'C3_Mean'),'high_sample'] <- 'C3'
#这里计算了各基因在所有样本中丰度的均值的平方根,用于作图时定义点的大小
EBSeq_stat$size <- apply(EBSeq_stat[1:3], 1, function(x) sqrt(mean(x)))
#ggtern 三元相图示例
library(ggtern)
tern_plot <- ggtern(EBSeq_stat, aes(C1_Mean, C2_Mean, C3_Mean)) +
geom_mask() +
geom_point(aes(color = high_sample, size = size), alpha = 0.6) +
scale_size(range = c(0, 4)) +
scale_colour_manual(values = c('red', 'blue', 'green3', 'purple', 'orange', 'skyblue', 'gray'), limits = c('C1', 'C2', 'C3', 'C1 & C2', 'C1 & C3', 'C2 & C3', 'none')) +
theme_bw() +
theme(axis.text = element_blank(), axis.ticks = element_blank()) +
labs(x = 'C1', y = 'C2', z = 'C3', color = 'en-riched') +
guides(size = FALSE)
#ggsave('tern_plot.pdf', tern_plot, width = 6.5, height = 5)
ggsave('tern_plot.png', tern_plot, width = 6.5, height = 5)
在这里,我们期望通过三元相图观测各基因更倾向于在哪些分组中富集。首先计算了各基因在哪个分组中的丰度最高;之后根据Pattern判别模式,结合基因具有最高丰度的分组,对这些基因作标记:它们更倾向于在哪些分组中富集;最后加载ggtern绘制三元图。作图结果如下所示。

关于更多的分组
虽然更多的分组EBSeq也能支持,但还是建议在比较时不超过4组数据吧。毕竟当分组很多时,如果你的数据量也大的话,不仅计算耗费资源,结果的解读及更进一步的分析也不是一件轻松的事情。
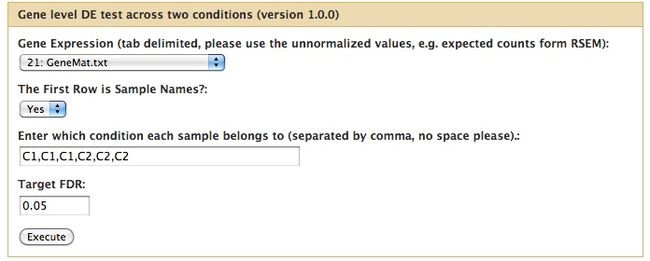
关于EBSeq图形化操作界面
EBSeq提供了图形化操作界面的R插件:https://www.biostat.wisc.edu/~ningleng/EBSeq_Package/EBSeq_Interface/,为一些相关的R代码,只不过当我们调用它们后即显示出图形界面,然后点击按钮操作就可以了,方便了不习惯命令行操作的同学们进行数据分析。
这个我就没再安装测试,有兴趣的同学们可以自己研究下。
除了R插件,EBSeq还提供了Galaxy插件,使得EBSeq同样可以在Galaxy平台上以图形化界面操作:https://www.biostat.wisc.edu/~ningleng/EBSeq_Package/EBSeq_Galaxy_toolshed/。有兴趣的同学们可以自行研究下。