蛋白质结构预测(swiss-model,modeller )
一、题目要求
MDEMATTQISKDELDELKEAFAKVDLNSNGFICDYELHELFKEANMPLPGYKVREIIQKLMLDGDRNKDG
KISFDEFVYIFQEVKSSDIAKTFRKAINRKEGICALGGTSELSSEGTQHSYSEEEKYAFVNWINKALEND
PDCRHVIPMNPNTDDLFKAVGDGIVLCKMINLSVPDTIDERAINKKKLTPFIIQENLNLALNSASAIGCH
VVNIGAEDLRAGKPHLVLGLLWQIIKIGLFADIELSRNEALAALLRDGETLEELMKLSPEELLLRWANFH
LENSGWQKINNFSADIKDSKAYFHLLNQIAPKGQKEGEPRIDINMSGFNETDDLKRAESMLQQADKLGCR
QFVTPADVVSGNPKLNLAFVANLFNKYPALTKPENQDIDWTLLEGETREERTFRNWMNSLGVNPHVNHLY
ADLQDALVILQLYERIKVPVDWSKVNKPPYPKLGANMKKLENCNYAVELGKHPAKFSLVGIGGQDLNDGN
QTLTLALVWQLMRRYTLNVLEDLGDGQKANDDIIVNWVNRTLSEAGKSTSIQSFKDKTISSSLAVVDLID
AIQPGCINYDLVKSGNLTEDDKHNNAKYAVSMARRIGARVYALPEDLVEVKPKMVMTVFACLMGRGMKRV
用swiss-model (3 models)和modeller (6 models)分别预测给定序列的结构,并用PROCHECK,Molprobity,Errat, Verify_3D分别评价所得预测模型,详细注释所得结果
依据打分情况,选出最佳的模型,说明理由。
建模注意事项:
了解本序列的结构组成(有几个结构域,分别是什么结构域……)
有针对性选择合适的模板进行建模
最后选出的最优模型用VMD显示三维结构,并注明每个结构域的位置
二、操作过程记录及结果
序列结构组成
为了对序列结构有个大体认识,现在uniprot中对序列进行BLAST,通过查找相似序列所对应蛋白质的结构,查看其结构域。发现人类Plastin-3蛋白质和给定序列Identity和Coverage都为100.0%,初步断定两者有相同的结构域,共计2个EF-hand,2个Actin-binding,然后每个Actin-binding都有两个亚基。

图表 1结构域
swiss-model建模
首先,进入swiss-model,输入序列,点击“Search For Templates”会单纯地进行模板搜索,搜索完毕后,根据需求,自己选择模板建模。点击“Build Model”会进行模板搜索,并自动选择模板建模,下面使用“Build Model”自动建模。
图表 2建模界面
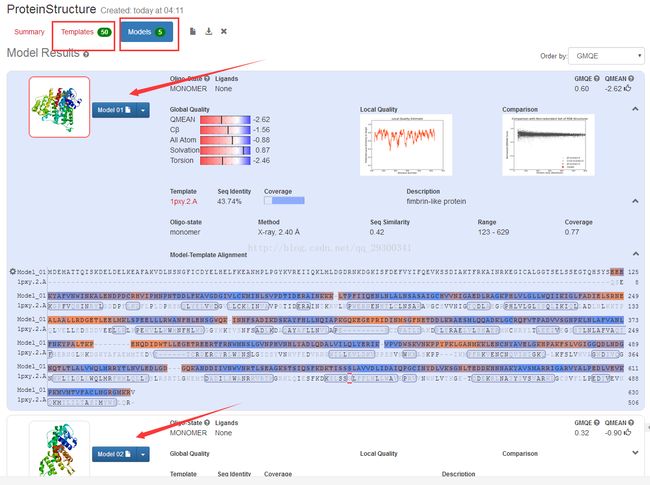
图表 3建模结果
可以看出来,总共搜出来50个Template模板,然后总共建出来5个Model。此处选择1个看上去漂亮的Model即可。
VMD可视化
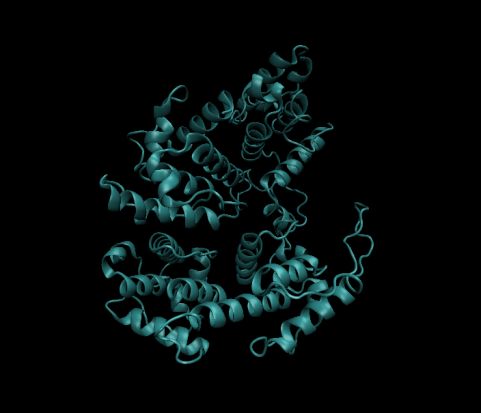
将模型下载为PDB格式,放进VMD可视化,整个蛋白质给人一种复杂精巧的感觉。(事实上是因为Swiss-model单模板建模,蛋白质不完整)
Modeller建模
Modeller教程:https://salilab.org/modeller/tutorial/basic.html
把以下两个教程中用到的文件下载下来,然后照着教程做一遍就可以大致明白了
https://salilab.org/modeller/tutorial/basic-example.zip
https://salilab.org/modeller/tutorial/advanced-example.zip
PS:Modeller安装后,会在开始菜单生成Modeller的命令框,在框内输入 “ mod9.18 脚本名称 ”即可。如果想运行速度更快,推荐python2.7,安装后"python 脚本名称"即可。
单模板建模
第一步、搜寻模板
Searching forstructures related to your protein sequence AND Selecting a template
搜寻模板、此步骤有很多种做法
1、 精致式做法:用Modeller的build_profile.py从本地序列数据库搜索,然后使用compare.py比较各个Template模板,得到一个distance matrix距离矩阵,然后根据距离矩阵,选择出想要的模板。(这种方法需要用到basic-example里面的pdb_95.pir,也可以在https://salilab.org/modeller/supplemental.html里面下载)
2、 粗放式做法:使用uniprot或者PDB等数据库的blast,搜出来相似序列,然后进入PDB找出心仪的序列,下载PDB结构,作为模板。
3、 简便式做法:直接使用swiss-model搜索出来的Template模板。
此处选择第三种方法,此种方法方便快捷,而且效果较好。
第二步、模板比对
Aligning yourprotein sequence with template
将自己的序列,按照basic-example文件夹中对应文件,改成ali格式,使用mod9.18运行align2d.py文件,生成myprotein-1aoaA.ali和myprotein-1aoaA.pap文件。
第三步、模型建立
Modelbuilding
模型建立,使用model-single.py,建立模型,总共生成六个模型

图表 4model-single结果
此处可以看出六个模板初步评估结果molpdf和DOPE score越低越好,GA341 score越接近1越好,这里选择第二个模型myprotein.B99990002.pdb
第四步、模型评估

图表 5单模板VMD可视化
使用VMD进行可视化,可以明显看出有大段区域没有进行建模,形成了三段尾巴,看出来单模板建模由于信息量有限,结果不好。
多模板建模
第一步、模板合并
首先使用salign.py进行模板间比对,从而合并各个模板。
第二步、模板比对
使用align2d_mult.py进行模板比对
第三步、手工添加模板
由于之前下载的多模板,从coverage可以看出来,都缺少开头的一段序列,因此进入swiss-model再拿开头的一段序列,寻找对应的模板2f2oA,然后使用单序列比对align2d.py,生成myprotein-2f2oA.ali比对结果文件,补充进myprotein-mult.ali
(注意,如果之前拿去Swiss-model搜模板是开头的那150个氨基酸,比对的时候myprotein最好用250个左右的氨基酸,从而使得插的空位是补在2f2oA序列上)
第四步、模型建立
使用model_mult.py建立模型

图表 6model_mult结果
第五步、模型可视化
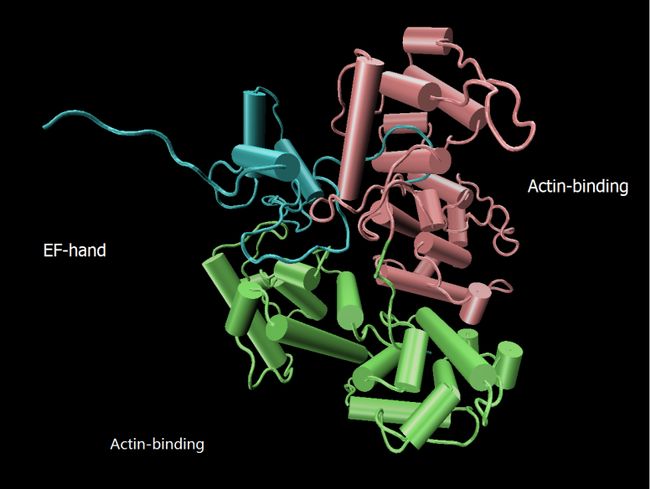
图表 7多模板VMD可视化
选择第三个模型进行可视化,毕竟是三个同源模板+一个补空位的模板,可视化效果很不错。
Modeller多模板建模深度优化
尝试进行Loop refining
首先,使用模型评价脚本evaluate_model.py,找到需要优化的区域,但是由于多模板建模没有生成几个模板合成的PDB文件,plot_profiles.py脚本不能用,自己用R语言绘图,然而由于无法绘制模板曲线,无法找到优化区域,此优化目前没有找到进行下去的办法
https://salilab.org/modeller/tutorial/advanced.html

使用model-segment.py脚本,指定用于建模的氨基酸长度,原来脚本也是不能简单地直接两个模板,我只能单纯地去掉前120个氨基酸,这样从原理上我就觉得还不如之前手动匹配氨基酸。
ModWeb在线建模
使用Modeller提供的在线服务,进行建模,改建模过程将花费较长时间,建议填写邮箱。
https://modbase.compbio.ucsf.edu/modweb/
模型细节

图表 8ModWeb模型细节
VMD可视化
选择第一个模型,由于是单模板,所以模板比较小,所以模型也就比较小,将模型下载为PDB格式,放进VMD可视化,这效果真的很棒,整个蛋白质就给人一种简洁而又不失精致、典雅而又不失自然的美感。
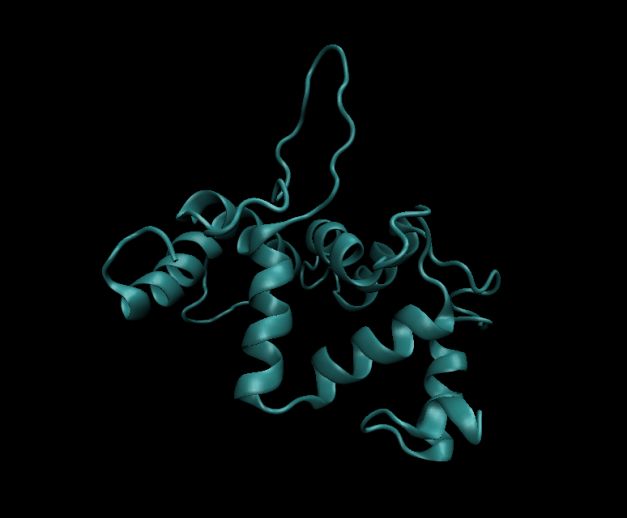
图表 9ModWeb的VMD可视化
模型评估
接下来,将对Swiss-model、Modeller多模板建模、ModWeb三个结果进行模型评估。
Swiss-model、Modeller多模板建模和ModWeb的VMD可视化结果可见上文。下面将使用PROCHECK、Molprobity、Errat、 Verify_3D进行评估
使用http://services.mbi.ucla.edu/SAVES/工具,相当于检测大礼包,可以一起运行PROCHECK、Errat和Verify_3D
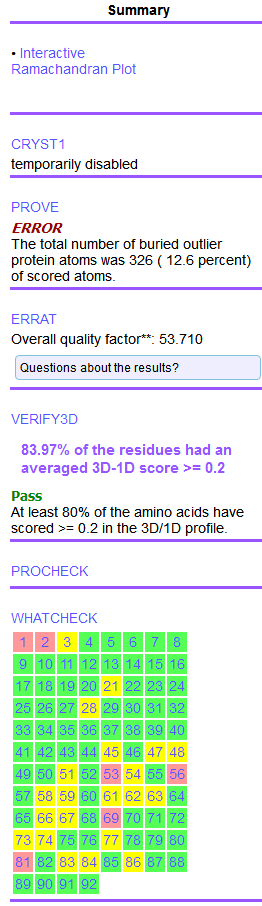
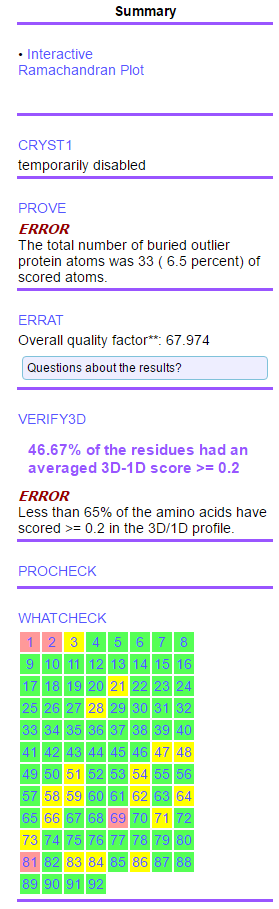
下图左图是Modeller多模板建模评分结果、中图是Swiss-model评分结果、右图是ModWeb评分结果。

PROCHECK是检测生理化学参数的,这个检测方式只有Swiss-model能评估,29个PASS,21个ERROR
VERIFY3D是检测3D结构的,Modeller多模板建模中83.97%的残基评分较高,Swiss-model建模中有92.06%的残基评分较高,ModWeb建模中有46.67%的残基评分较高,此项评估Swiss-model胜出。
Errat是根据结晶学评估蛋白质三维结构的,此时Swiss-model建模中得分为93.555,Modeller多模板建模得分为53.710,ModWeb评分为67.974,此项评估Swiss-model胜出。
Molprobity评估较为严格,需要经过加氢等一系列步骤,Swiss-model和ModWeb都有绿色较好的项,而Modeller最好的也是一个橙色的警告项,此项评估Swiss-model胜出。
Errat评估细节
黑线多就是错误率高,这项明显Swiss-model黑线要少得多,更好一些。
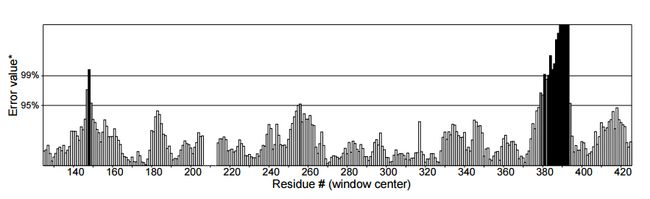
图表 10Swiss-model的Errat评分
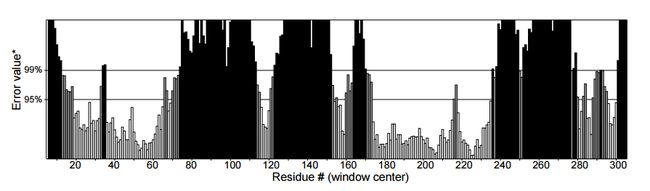
图表 11Modeller的Errat评分
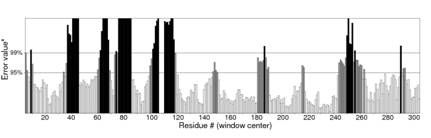
图表 12ModWeb的Errat评分
PROCHECK评估细节
下面是拉曼图(Ramachandran)左图是Swiss-model建模结果、中图是Modeller多模板结果,右图为ModWeb结果。
黄色为允许区域,红色为最大允许区域,白色为不允许区域。两者红色和黄色区域较为一致。
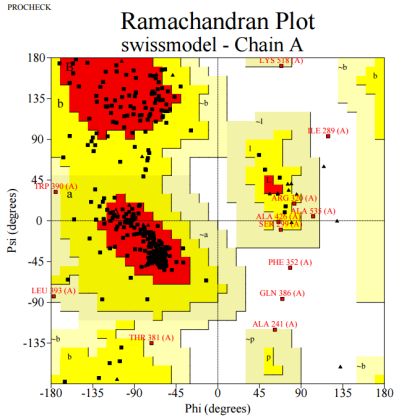

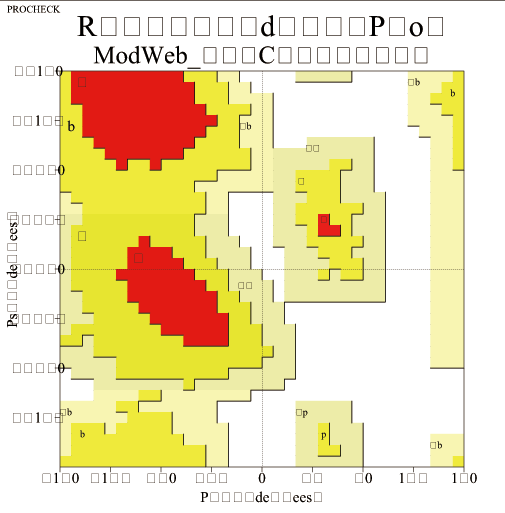
Molprobity评估细节
Molprobity会经过加氢等步骤,先对PDB文件进行修改,再评估,绿色为很好,橙色为警告,红色为不可接受。

图表 13 Swiss-model的Molprobity评分

图表 14 Modeller多模板的Molprobity评分
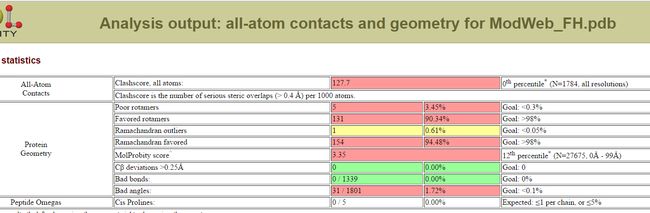
图表 15ModWeb的Molprobity评分
三、分析与讨论
首先对VMD可视化结果大体观察,Swiss-model和Modeller看上去都蛮复杂,感觉像是有EF-hand和Actin-binding,其中Actin-binding大概也是有两个亚基,而ModWeb可视化后,实在是有点过于简单,看不出有这么复杂的结构,Swiss-model和Modeller多模板建模更胜一筹。
使用Swiss-model在线建模,Modeller多模板本地建模,Modeller在线服务器ModWeb建模,三者进行比较,可以发现Modeller本地建模和在线服务器ModWeb评分不相上下各项指标互有高低,多项指标Modeller比ModWeb要高,Swiss-model在线建模效果评分最好。
综上,目前Swiss-model建模效果评分最高,但是本质上Swiss-model和ModWeb都是单模板建模,没有成功建模的部分都没有加上去,所以我最后选择自己多模板建立的模型,并且可视化结构域。如果Modeller本地建模能继续优化,使用examples/automodel/model-segment.py指定各部分的模板,还能做得更好。
https://salilab.org/modeller/9.18/manual/node23.html
PS:做作业的时候发现13级学姐的帖子,大家也可以去参考一下:http://www.dxy.cn/bbs/topic/31372707