今天开始会写一系列关于用R进行基因芯片分析的文章,算是对该流程的总结。之前学习了通过circos来画圈图,以及通过MCScanX进行基因家族的共线性分析,放一张结果先:
逼格满满,容我先傲娇一下~
##################我是分割线##################
GEO数据库基础知识
Gene Expression Omnibus(简称GEO)是一个将科研团体提交的基因芯片、下一代测序数据、以及其他高通量功能基因组数据形式进行归档以供免费下载的公共数据库。
在GEO中记录的数据形式主要有一下几种:
- GEO Platform (GPL) 平台,包括芯片和测序平台,每个平台都被赋予唯一的GEO号
- GEO Series (GSE) study的ID号,是相关实验样本的集合,并提供了对于该研究的关键描述性信息
- GEO Sample (GSM) 样本ID号,提供了单个样本处理的实验条件以及该样本的测量数据
- GEO Dataset (GDS) 数据集的ID号
一个GDS可以有多个GSM,一个GSM可以有多个GSE,至于GPL,一般不接触。
简单来讲,分析一张表达芯片主要包括以下几个步骤:
1. 安装相关的packages(installation)
2. 从GEO下载数据(download dataset)
3. 归一化(normalization)
4. 芯片质控(quality control)
5. 差异表达基因分析(differential expressed gene analysis)
我们通常接触的都是GSE系列(一个GSE里面有多个GSM)的数据。
Installation
从GEO数据库中下载感兴趣的芯片,需要事先安装好bioconductor的核心包,然后安装专门用于下载标准GEO数据结构的GEOquery包。
R console 输入以下命令,进行GEOquery包的安装:
source("http://bioconductor.org/biocLite.R")
biocLite("GEOquery")
Getting the data
这次以数据集GSE97045为对象进行分析。这张芯片以栽培番茄(S. lycopersicum (Sl) (cv. P73) )和野生潘那利番茄(S. pennellii (Sp) (acc. PE47) )为研究对象,分析干旱处理前后,两种番茄种内与种间基因表达的差异,并进行聚类分析。可以发现,该实验包括两个因素:不同番茄(栽培 vs 野生)和不同处理(unstressed vs drought)。每一条件各有三个重复,因此,该实验的总样本(芯片)数为:2(两种番茄)* 2 (两个处理)* 3 (三次重复)= 12
首先,通过GEOquery包进行芯片数据的下载或直接在本地打开文件:
setwd('D:\\work\\Bioinformatics\\data\\array')
gse97045 <- getGEO(filename = "D:\\work\\Bioinformatics\\data\\array\\GSE97045_series_matrix.txt.gz", getGPL = F)
#class(gse97045)
#[1] "ExpressionSet"
#attr(,"package")
#[1] "Biobase"
直接打开的就是"ExpressionSet"类文件,可以用该类的方法,主要包括4种:
#geneNames(): 该方法已失效
#sampleNames(): 返回样本(芯片)编号
> sampleNames(gse97045)
[1] "GSM2550523" "GSM2550524" "GSM2550525" "GSM2550526" "GSM2550527" "GSM2550528" "GSM2550529" "GSM2550530" "GSM2550531" "GSM2550532" "GSM2550533" "GSM2550534"
#pData(): 返回与实验相关的表型数据或元数据,即phenotypic data的缩写,以data.frame的格式呈现
> colnames(pData(gse97045))
[1] "title" "geo_accession" "status"
[4] "submission_date" "last_update_date" "type"
[7] "channel_count" "source_name_ch1" "organism_ch1"
[10] "characteristics_ch1" "characteristics_ch1.1" "characteristics_ch1.2"
[13] "characteristics_ch1.3" "treatment_protocol_ch1" "growth_protocol_ch1"
[16] "molecule_ch1" "extract_protocol_ch1" "label_ch1"
[19] "label_protocol_ch1" "taxid_ch1" "hyb_protocol"
[22] "scan_protocol" "description" "data_processing"
[25] "data_processing.1" "platform_id" "contact_name"
[28] "contact_email" "contact_laboratory" "contact_department"
[31] "contact_institute" "contact_address" "contact_city"
[34] "contact_state" "contact_zip/postal_code" "contact_country"
[37] "supplementary_file" "data_row_count" "age:ch1"
[40] "condition:ch1" "cultivar/accession:ch1" "tissue:ch1"
看一下元数据的“title”列信息,告诉我们样本信息、处理信息、以及重复信息,factor格式数据:
> pData(gse97045)$title
[1] Tomato (S. lycopersicum) cv. P73, control condition, rep. 1
[2] Tomato (S. lycopersicum) cv. P73, control condition, rep. 2
[3] Tomato (S. lycopersicum) cv. P73, control condition, rep. 3
[4] Tomato (S. lycopersicum) cv. P73, water stress condition, rep. 1
[5] Tomato (S. lycopersicum) cv. P73, water stress condition, rep. 2
[6] Tomato (S. lycopersicum) cv. P73, water stress condition, rep. 3
[7] S. pennellii acc. PE47, control condition, rep. 1
[8] S. pennellii acc. PE47, control condition, rep. 2
[9] S. pennellii acc. PE47, control condition, rep. 3
[10] S. pennellii acc. PE47, water stress condition, rep. 1
[11] S. pennellii acc. PE47, water stress condition, rep. 2
[12] S. pennellii acc. PE47, water stress condition, rep. 3
12 Levels: S. pennellii acc. PE47, control condition, rep. 1 ...
#exprs(): Retrieve expression data from eSets.
> head(exprs(gse97045))
GSM2550523 GSM2550524 GSM2550525 GSM2550526 GSM2550527 GSM2550528
AFFX-BioB-3_at 9.154191 8.935244 8.765674 8.839061 8.275339 8.609382
AFFX-BioB-5_at 9.115401 8.814133 8.664128 8.730531 8.321579 8.564144
AFFX-BioB-M_at 8.980967 8.748333 8.642012 8.750143 8.249259 8.606312
AFFX-BioC-3_at 10.598291 10.439536 10.294270 10.344476 9.912000 10.224729
AFFX-BioC-5_at 10.409473 10.211119 10.013224 10.126247 9.690638 9.981757
AFFX-BioDn-3_at 12.484206 12.394027 12.261628 12.482427 12.047057 12.336597
GSM2550529 GSM2550530 GSM2550531 GSM2550532 GSM2550533 GSM2550534
AFFX-BioB-3_at 9.477754 9.479547 9.513700 9.179431 9.239031 9.238477
AFFX-BioB-5_at 9.402628 9.429561 9.535614 9.166966 9.273455 9.244174
AFFX-BioB-M_at 9.405048 9.328257 9.393749 9.061191 9.068758 9.223863
AFFX-BioC-3_at 10.878668 10.895548 10.923604 10.612944 10.669775 10.707216
AFFX-BioC-5_at 10.588091 10.621175 10.646094 10.341966 10.442737 10.434916
AFFX-BioDn-3_at 12.603844 12.596163 12.627703 12.487721 12.563084 12.577017
这里再插点有关exprs()函数的知识:
- exprs()属于Biobase package,专门用于处理eSet类数据,返回值为基因表达矩阵。
- eSet类是Bioconductor 为基因表达数据格式所制定的标准,基本可称为一个Meta类,也是一个虚类,衍生出了许多重要的类,包括ExpressionSet类,SnpSet类以及AffyBatch类等1。
将该过程包装为函数:
downGSE <- function(studyID, destdir = ".") {
library(GEOquery)
eSet <- getGEO(studyID, destdir = destdir, getGPL = F)
exprSet <- exprs(eSet[[1]])
pdata <- pData(eSet[[1]])
write.csv(exprSet, paste0(studyID, "_exprSet.csv"))
write.csv(pdata, paste0(studyID, "_metadata.csv"))
return(eSet)
}
对GSE97045进行作图,看其是否已经归一化:
> library(affyPLM)
载入需要的程辑包:gcrma
载入需要的程辑包:preprocessCore
> boxplot(gse97045, col = cols)
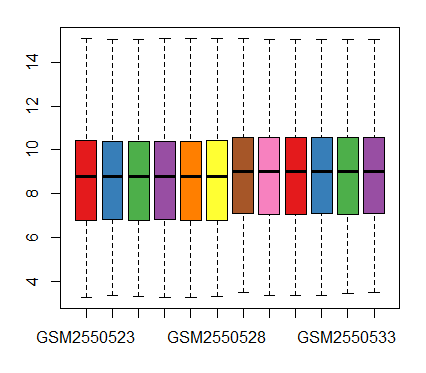
可见,下载的GSE97045_series_matrix.txt.gz是经过归一化处理的表达矩阵,可以直接应用于下游数据分析。
Reference:
- 高山, 欧剑虹,肖凯,《R语言与Bioconductor生物信息学应用》