整合QC质控结果的利器——MultiQC
一、MultiQC介绍
NGS技术的进步催生了新的实验设计、分析类型和极高通量测序数据的生成。对于这些数据的质量评估,每一步分析结果的评估是后续结果可信度的衡量和保障。不少生信工具都可以给样品生成一个评估结果,如FastQC、Qualimap 和RSeQC等 (39个转录组分析工具,120种组合评估)。但是这时又出现了一个难题,那就是几乎所有的质控工具都是针对单个样本生成一个报告,这就要求用户自己去逐一查找各个QC结果,这无疑是个十分耗时、重复又复杂的事,而且还不能快速看出所有样本的异同。
那能否把所有质控结果整合在一起呢?可以自己写程序造轮子(我们之前就是这么做的)。但现在有了MultiQC,基于Python的小工具很好地解决了这个繁琐的事情,其强大的功能主要体现在以下三个方面:
1)能将测序数据的多个QC结果整合成一个HTLM网页交互式报告,同时也能导出pdf文件;
2)支持多种分析类型的质控结果查看,如:RNAseq、Whole-Genome Seq、Bisulfite Seq、Hi-C和MultiQC_NGI;
3)支持整合68种软件分析的结果,而且支持的软件还在持续增加,也可以自己写作一个插件,具体见下图。

二、安装MultiQC
依赖python2.7+, 3.4+ 或者 3.5+
# pip安装
pip install git+https://github.com/ewels/MultiQC.git #Installation with pip
# conda安装
conda install -c bioconda multiqc # Installing with conda
三、运行MultiQC
直接指定MultiQC要分析的文件路径即可,若数据在当前目录下输入multiqc .即可。
multiqc .
multiqc data/
multiqc data/ ../proj_one/analysis/ /tmp/results
multiqc data/*_fastqc.zip
multiqc data/sample_1*
使用--ignore忽略掉某些文件
multiqc . --ignore *_R2*
multiqc . --ignore run_two/
multiqc . --ignore */run_three/*/fastqc/*_R2.zip
四、MultiQC报告解读(以RNA-Seq数据为例)
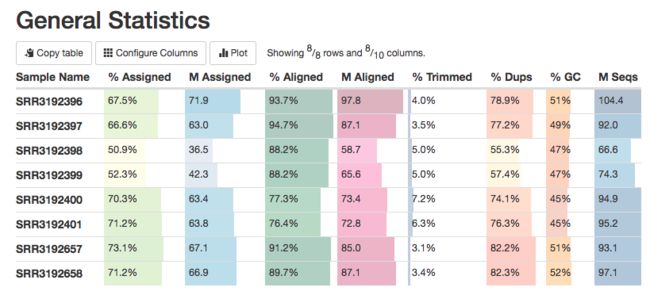
1. General Statistics
每一个样本reads数量、比对层面的质量评估整合统计表,点击Configure Columns可以选择显示或不显示某些项。点击Plot可以绘图。
点击Configure Columns选择展示哪些项

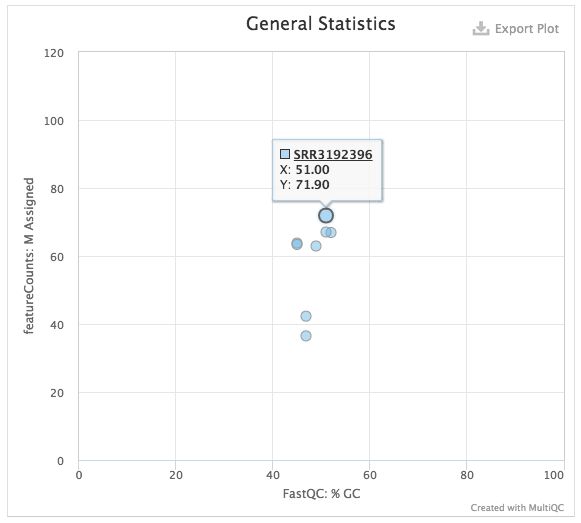
点击Plot可以对任意两种属性的评估结果做交互式二维图,若各样本均一性好,散点会比较集中,反之会出现某些离散的点,这样方便查看某些指标异常的离群样本。
2. featureCounts
利用featureCounts工具计算每个基因外显子的reads数的结果展示。featureCounts不仅可以支持gene的定量,也支持exon, gene bodies, genomic bins, chromsomal locations的定量。功能类似的软件是HTSeq。
软件官网:http://bioinf.wehi.edu.au/featureCounts/

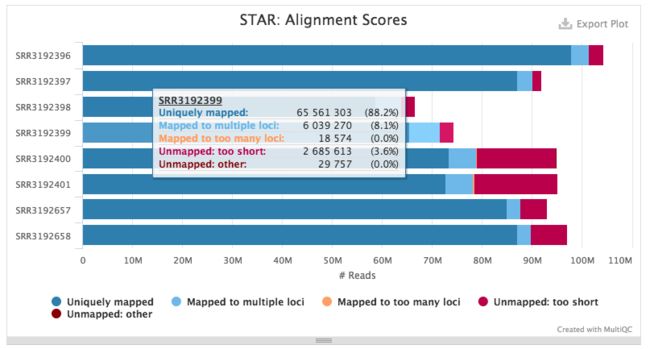
3. STAR
基于STAR比对工具的分析结果,STAR会将没有paired mapping的reads都剔除,避免single reads比对到基因组上;并且STAR对lower-quality(采用more soft-clipped和错配碱基)比对有较高的容忍度。
软件官网:https://github.com/alexdobin/STAR
更多分析工具比较见:转录组分析工具大比拼
4. Cutadapt
用cutadapt软件来对双端测序数据去除接头后的结果。
对测序数据进行过滤时cutadapt对测序数据进行识别、剪切并去除adapters, primers , poly_A等序列,移除被adapter污染的reads部分(指由于插入片段长度不够,测序仪读到的测序引物等序列)。具体见NGS基础 - 高通量测序原理。
软件官网:https://cutadapt.readthedocs.io/en/stable/

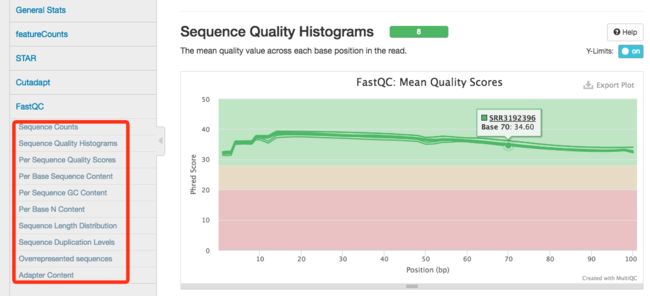
5. FastQC
MultiQC将fastqc工具分析得到的10个结果分别整合成一个模块,集中查看。
软件官网:http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
具体的关于FastQC报告解读可以见历史推文:NGS基础 - FASTQ格式解释和质量评估
MultiQC的可定制性也比较强,更多功能可以继续探索。