话休絮烦
主要流程:
- 将reads比对到基因组上;
- 将比对好的reads进行拼装;
- 量化基因的表达水平;
- 计算在不同条件下的差异表达分析

原始数据:ftp://ftp.ccb.jhu.edu/pub/RNAseq_protocol,后续都是对此数据进行分析的。
这个protocol首先从原始RAN-seq数据入手,输出:基因list,转录本,每个样本的表达量,能够表现差异表达基因的表格,显著性的计算。
使用HISAT将读段 reads 匹配到参考基因组上,使用者可以提供注释文件,但HISAT依旧会检测注释文件没有列出来的剪切位点;
比对上的reads将会被呈递给StringTie进行转录本组装,StringTie单独的对每个样本进行组装,在组装的过程中顺带估算每个基因及isoform的表达水平。
所有的转录本都被呈递给StringTie的merge函数进行merge,这一步是必须的,因为有些样本的转录本可能仅仅被部分reads覆盖,无法被第二步的StringTie组装出来。merge步骤可以创建出所有样本里面都有的转录本,方便下一步的对比。
4. merge的数据再一次被呈递给StringTie,StringTie可以利用merge的数据重新估算转录本的丰度,还能额外的提供转录本reads数量的数据给下一步的ballgown
- 最后一步:Ballgown从上一步获得所有转录本及其丰度,根据实验条件进行分类统计。
前期准备:软件安装
在按着paper操作之前建议先将软件bing/google一下,了解相关的安装等操作。可以自行将conda,R,以及相关的R的包搜索一下。
安装软件,我是用conda进行安装的:
$ conda install hisat2$ conda install stringtie$ conda install samtools
安装自己的R:
$ conda search R`
$ conda install -c r r`
$ export R_LIBS=$HOME/Rlib64`
R包安装:**Ballgown **(ps: 最近的R包安装有改动,按照文档上安装也可以)
$ R
> install.packages("devtools",lib="Rlib64",repos="https://mirrors.tuna.tsinghua.edu.cn/CRAN/")
> source("http://www.bioconductor.org/biocLite.R")
> biocLite(c("alyssafrazee/RSkittleBrewer","ballgown","genefilter","dplyr","devtools"))
数据处理设计
Read alignment with HISAT
总体上来说HISAT利用了BWA和Bowtie的算法,同时解决了mRNA中不存在内含子难以比对的问题,比对上代主流RNA-seq比对工具能快50倍,同时需求更少的内存<8G
参考运行的命令如下:
$ hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran -1 chrX_data/samples/ERR188044_chrX_1.fastq.gz -2 chrX_data/samples/ERR188044_chrX_2.fastq.gz -S ERR188044_chrX.sam
paper中有每个样本的命令

后面具体的命令详见文章中的PROCEDURE。
用Stringtie进行转录本组装量化
RNA-seq的分析依赖于精准的对于基因isoform的重建以及对于基因相对丰度的预测。继承于Cufflinks,StringTie相对于之前开发的工具更为准确,需求内存和耗时也更少。使用者一样可以使用注释文件来帮助StringTie运行,对于低丰度的数据比较有帮助。此时StringTie依旧会对非注释区域进行转录本的组装,所以注释文件在这里是可选选项。命令行如下:
$ samtools sort -@ 8 -o ERR188044_chrX.bam ERR188044_chrX.sam
$ stringtie -p 8 -G chrX_data/genes/chrX.gtf -o ERR188044_chrX.gtf –l ERR188044 ERR188044_chrX.bam
转录本组装完成后,使用者可以利用gffcompare(StringTie工具包含)工具来得知有多少转录本和注释文件相同,又有多少新的转录本.
#input: gff or gtf format
transcripts.gtf
commandline example:
$ gffcompare -G -r chrX.gtf transcripts.gtf
#-r : reference
#-G :compare all transcripts
#-o :prefix
output file 1: gffcmp.annotated.gtf
# 显示StringTie组装的转录本与注释文件内的转录本的差别
output dile 2: gffcmp.stats
# 根据注释文件显示组装结果的准确性和阳性预测率
用Ballgown进行差异表达
组装和定量完转录本之后自然需要进行基因表达差异分析,统计建模和可视化。R和Bioconductor提供了一揽子的工具处理这些任务,包括raw data的plot作图,数据的标准化,下游的统计建。此处使用Ballgown包作为承上启下的桥梁。
在R里面使用Ballgown处理需要得到:
- 表型数据。关于样本的信息
- 表达数据。标准化和未标准化的关于外显子,junction,转录本,基因的表达数量
- 基因信息。有关外显子,junction,转录本,基因的坐标以及注释信息
而大多数的差异表达分析都会包括下面几个步骤:
- 数据可视化和检查
- 差异表达的统计分析
- 多重检验校正
- 下游检查和数据summary
Ballgown包可以完成以上的几个步骤并且可以联合R语言的其它操作进行分析
Ballgown使用:
- 数据的读入:需要将StringTie输出的数据结合表型数据,这里需要保证表型数据的identifier和StringTie输出数据的一致,不然会报错
- 预测丰度的检查:以FPKM(fragments per kilobase of transcript per million mapped reads)为单位的丰度预测将会根据library size进行标准化。#极端差异此处需要引起注意
- 使用线性模型进行差异表达分析,由于FPKM对于转录本解读过于曲解,所以这里需要使用log转化处理数据,随后再使用线性模型进行差异分析。
- Ballgown可以对time-course和fixed-condition数据进行差异分析,但是无法避免批次效应带来的误差。#使用stattest功能可以指定任何已知的cofounder
- Ballgown可以帮助你在基因,转录本,外显子,junction水平上进行差异分析
- 结果会以表格形式展现,如果样本够多还有p值和q值
- 结果数据可以用来进行下一步的gene set分析(例如GSEA)等等
实战示例
准备阶段
实际上本轮操作就是按照文章给的示例数据走了一遍流程:
文章中,为了减少计算时间,方便读者重复,作者截取了一批实验数据中能够匹配到chromosomeX上的数据作为示例数据,chromosomeX是一个基因相对较为丰富的染色体,可以占到基因组的5%左右。
首先是下载数据随后解压:
$nohup wget ftp://ftp.ccb.jhu.edu/pub/RNAseq_protocol/chrX_data.tar.gz 2>download.log &
$tar zxvf chrX_data.tar.gz
数据如下:
ps:解释一下geuvadis_phenodata.csv,他是每个sample的样本信息,从两地人群中(英格兰岛住民和约鲁巴住民)各取六个样,六个样又分为男女性别各三个(最少的生物学重复)。信息内容如下:
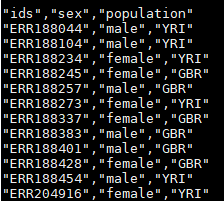
ids 代表是样本,sex和population是样本标签。
随后介绍一个骚命令:
HISAT2可以直接从NCBI下载sra格式的文件并作为输入文件格式
下面以ERR188245测序数据为例
$ hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran --sra-acc ERR188245 –S ERR188245_chrX.sam
可以说是相当的帮人偷懒了
在Hisat2 中的 Index文件可以从https://ccb.jhu.edu/software/hisat2/index.shtml 网站上下载下来。
如果需要建立官网上没有基因组的index,就需要需要使用Hisat2包里面的python脚本:
extract_splice_sites.py和extract_exons.py,从注释文件里面抽取出剪切位点和外显子信息
以后的步骤可以按文章中的一步步进行:
^Align the RNA-seq reads to the genome ● TIMING <20 min
1| Map the reads for each sample to the reference genome:
$ hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran -1 chrX_data/samples/ERR188044_chrX_1.fastq.gz -2 chrX_data/samples/ERR188044_chrX_2.fastq.gz -S ERR188044_chrX.sam
2| Sort and convert the SAM files to BAM:
$ samtools sort -@ 8 -o ERR188044_chrX.bam ERR188044_chrX.sam
samtools 1.8(conda)运行时出现问题,后来用系统的/usr/local/bin/samtools来解决
$ samtools
samtools: error while loading shared libraries: libtinfow.so.5: cannot open shared object file: No such file or directory
ln -s /usr/lib/libncurses.so.5 /lib64/libtinfo.so.5
ln -s /usr/lib/libncurses.so.5 /usr/lib/libtinfo.so.5
^Assemble and quantify expressed genes and transcripts ● TIMING ~15 min
3| Assemble transcripts for each sample:
$ stringtie -p 8 -G chrX_data/genes/chrX.gtf -o ERR188044_chrX.gtf -l ERR188044 ERR188044_chrX.bam
4| Merge transcripts from all samples:
$ stringtie --merge -p 8 -G chrX_data/genes/chrX.gtf -o stringtie_merged.gtf chrX_data/mergelist.txt
5| Examine how the transcripts compare with the reference annotation (optional):
$ gffcompare -r chrX_data/genes/chrX.gtf -G -o merged stringtie_merged.gtf
the -o option specifies the prefix to use for output files that gffcompare will create.
6| Estimate transcript abundances and create table counts for Ballgown:
$ stringtie -e -B -p 8 -G assembled_reads/stringtie_merged.gtf -o ballgown/ERR188044/ERR188044_chrX.gtf mapped_reads/ERR188044_chrX.bam
^Run the differential expression analysis protocol ● TIMING ~5 min
7| Load relevant R packages.
$ R
library(ballgown)
library(RSkittleBrewer)
library(genefilter)
library(dplyr)
library(devtools)
8| Load the phenotype data for the samples. An example file called geuvadis_phenodata.csv is included with the
data files for this protocol (ChrX_data).
pheno_data = read.csv("chrX_data/geuvadis_phenodata.csv")
9| Read in the expression data that were calculated by StringTie. To do this, we use the ballgown command with the following three parameters: the directory in which the data are stored (dataDir, which here is named simply ‘Ballgown’), a pattern that appears in the sample names (samplePattern) and the phenotypic information that we loaded in the previous step (pData). Here we present a standardized pipeline that can be used to perform standard differential expression analysis.
bg_chrX = ballgown(dataDir = "ballgown", samplePattern = "ERR", pData=pheno_data)
10| Filter to remove low-abundance genes. Another approach that has been used for gene expression analysis is to apply a variance filter. Here we remove all transcripts with a variance across samples less than one:
bg_chrX_filt = subset(bg_chrX,"rowVars(texpr(bg_chrX)) >1",genomesubset=TRUE)
11| Identify transcripts that show statistically significant differences between groups. As an example, we will look for transcripts that are differentially expressed between sexes, while correcting for any differences in expression due to the population variable. We set the getFC=TRUE parameter so that we can look at the confounder-adjusted fold change between the two groups.
For small sample sizes (n < 4 per group), it is often better to perform regularization. This can be done using the limma package in Bioconductor. The statistical test uses a cumulative upper quartile normalization.
results_transcripts = stattest(bg_chrX_filt, feature="transcript", covariate="sex", adjustvars = c("population"), getFC=TRUE, meas="FPKM")
12| Identify genes that show statistically significant differences between groups. For this we can run the same function that we used to identify differentially expressed transcripts, but here we set feature="gene" in the stattest command:
results_genes = stattest(bg_chrX_filt, feature="gene", covariate="sex", adjustvars = c("population"), getFC=TRUE, meas="FPKM")
13| Add gene names and gene IDs to the results_transcripts data frame:
results_transcripts = data.frame(geneNames=ballgown::geneNames(bg_chrX_filt), geneIDs=ballgown::geneIDs(bg_chrX_filt), transcriptNames=ballgown::transcriptNames(bg_chrX_filt), results_transcripts)
14| Sort the results from the smallest P value to the largest:
results_transcripts = arrange(results_transcripts,pval)
results_genes = arrange(results_genes,pval)
15| Write the results to a csv file that can be shared and distributed:
write.csv(results_transcripts, "chrX_transcript_results.csv", row.names=FALSE)
write.csv(results_genes, "chrX_gene_results.csv", row.names=FALSE)
Table 3 | Differentially expressed transcripts between sexes (q value <5%).
16| Identify transcripts and genes with a q value <0.05:
subset(results_transcripts,results_transcripts$qval<0.05)
subset(results_genes,results_genes$qval<0.05)
As shown in the tables, chromosome X has nine transcripts that are differentially expressed between the sexes (using a q value threshold of 0.05), three of which correspond to isoforms of known genes (XIST, TSIX and PNPLA4).
Table 4 | Differentially expressed genes between sexes (q value <5%).
^Data visualization ● TIMING variable
You can use Ballgown to visualize RNA-seq results in a variety of ways. The plots produced by Ballgown make it easier to view and compare expression data, and some commands can generate publication-ready figures.
17| Make the plots pretty. This step is optional, but if you do run it you will get the plots in the nice colors that we used to generate our figures:
tropical= c('darkorange', 'dodgerblue', 'hotpink', 'limegreen', 'yellow')
palette(tropical)
18| Show the distribution of gene abundances (measured as FPKM values) across samples, colored by sex (Fig. 3).
Ballgown stores a variety of measurements that can be compared and visualized. For this protocol, we will compare the FPKM measurements for the transcripts, but you can also obtain measurements for each splice junction, exon and gene in the data set. The first command below accesses the FPKM data. The plots will be easier to visualize if you first transform the FPKM data; here we use a log2 transformation that adds one to all FPKM values because log2(0) is undefined (second command below). The third command actually creates the plot:
fpkm = texpr(bg_chrX,meas="FPKM")
fpkm = log2(fpkm+1)
tiff(filename="fpkm.tiff")
boxplot(fpkm,col=as.numeric(pheno_data$sex),las=2,ylab='log2(FPKM+1)')
dev.off()
Figure 3 | Distribution of FPKM values across the 12 samples. Samples
from the same sex are shown in the same color: males in blue, and females
in orange.
19| Make plots of individual transcripts across samples. (Fig. 4). The first two commands below show the name of the transcript (NM_012227) and the name of the gene that contains it (GTP binding protein 6, GTPBP6):
ballgown::transcriptNames(bg_chrX)[12]
ballgown::geneNames(bg_chrX)[12]
tiff(filename="fpkm_transcript.tiff")
plot(fpkm[12,] ~ pheno_data$sex, border=c(1,2), main=paste(ballgown::geneNames(bg_chrX)[12],' : ', ballgown::transcriptNames(bg_chrX)[12]), pch=19, xlab="Sex", ylab='log2(FPKM+1)')
points(fpkm[12,] ~ jitter(as.numeric(pheno_data$sex)), col=as.numeric(pheno_data$sex))
dev.off()
Figure 4 | FPKM distributions in males and females for transcript NM_012227
from gene GTPBP6 (GTP binding protein 6), a gene that is known to be more highly expressed in males, displayed as box-and-whiskers plots.
20| Plot the structure and expression levels in a sample of all transcripts that share the same gene locus. For example, in Figure 5 we show all the transcripts from the gene that contains the first transcript, NR_001564, in Table 3. This is the 1,729th transcript in the Ballgown object according to the id column in the table, and we can easily find the gene ID of the transcript by using ballgown::geneIDs(bg_chrX)[1729]. This reveals that NR_001564 is an isoform of the gene XIST, which is known to be much more highly expressed in females than in males. We can see in Table 3 that other isoforms of this gene appear to be differentially expressed as well. We can plot their structure and expression levels by passing the gene name and the Ballgown object to the plotTranscripts function. The plot shows one transcript on each row, colored by its FPKM level. By default, the transcripts at that locus are colored by their expression levels in the first sample, but we can tell the plotTranscripts function which sample we are interested in—here we choose sample ERR188234:
tiff(filename="gene_expression.tiff")
plotTranscripts(ballgown::geneIDs(bg_chrX)[1729], bg_chrX, main=c('Gene XIST in sample ERR188234'), sample=c('ERR188234'))
dev.off()
Figure 5 | Structure and expression levels of five distinct isoforms of the XIST gene in sample ERR188234. Expression levels are shown in varying shades of yellow. The third isoform is expressed at a much higher level than the others, as indicated by the darker color.
21| We can also plot the average expression levels for all transcripts of a gene within different groups using the plotMeans function. We need to specify which gene to plot, which Ballgown object to use and which variable to group by. As an example, plot the second gene in Table 4, MSTRG.56, using the following command.
tiff(filename="plotMeans.tiff")
plotMeans('MSTRG.56', bg_chrX, groupvar="sex",legend=FALSE)
dev.off()
前面的重复命令我只列了一行,大家按照paper上的命令运行即可.
文章转自:RNA-seq数据分析---方法学文章的实战练习