蛋白质结构预测与分析
蛋白质的结构
一级结构
氨基酸序列
二级结构
周期性的结构构象
三级结构
整条多肽链的三维空间结构
四级结构
多个亚基形成的复合体结构
蛋白质的二级结构
蛋白质经过折叠后会形成规则的片段,这些规则的片段构成了蛋白质的二级结构单元。
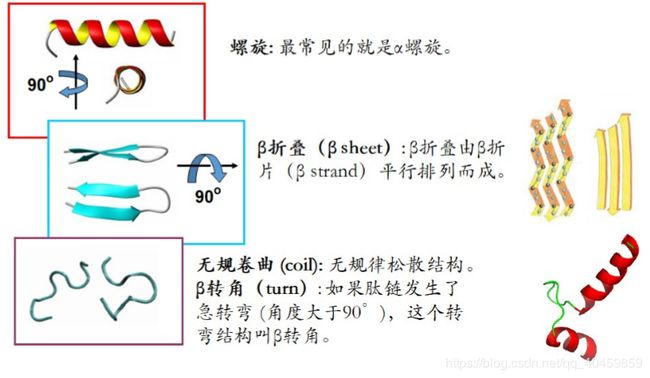 图 1蛋白质的二级结构
图 1蛋白质的二级结构
DSSP指认
DSSP(Definition of Secondary Structure of Proteins),即蛋白质二级结构定义词典。DSSP并不预测二级结构,而是根据二级结构的定义对已经测定三级结构的蛋白质的各个位置指认出是哪种二级结构。
PDB获取
PDB 里没有现成的针对某一个蛋白质的 FASTA 格式二级结构序列下载链接。“Download FASTA File”链接只能下载 FASTA 格式的一级结构序列,也就是氨基酸序列。
软件预测
已知空间结构的蛋白质在 PDB 数据库里只有 10 万多个。然而,UniprotKB 数据库里却有几百万条蛋白质序列。也就是说,绝大多数蛋白质的空间结构还都未知。这些蛋白质的二级结构(α螺旋、β折片)需要用计算机软件来预测。
蛋白质的三级结构
通过搜索PDBID、分子名称、作者姓名等关键词获取蛋白质三级结构。
通过序列相似性搜索获得与输入序列同源的蛋白质三级结构。
PDB 文件是通过记录蛋白质中每一个氨基酸上的每一个原子的三维坐标来存储空间结构信息的。这些原子坐标可以被三维可视化软件读取。三维可视化软件能够创建一个三维空间,然后根据原子坐标以及原子的大小把原子展示在空间内,并根据原子间的距离给它们连上化学键。这样一个立体的蛋白质结构就呈现在眼前了。
三级结构可视化软件VMD
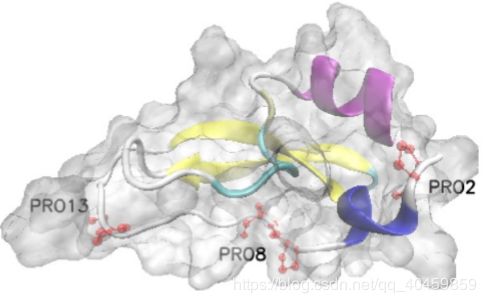 图 2 最终结构图
图 2 最终结构图
计算方法预测三级结构
同源建模法(SWISS-MODEL)
原理:相似的氨基酸序列对应着相似的蛋白质结构。
- 找到与目标序列同源的已知结构作为模板。
- 为目标序列与模板序列创建序列比对。
- 根据创建的序列比对,用同源建模软件预测结构模型。
- 评估模型质量,并根据评估结构重复以上过程,直至模型质量合格。
预测效果:如果目标序列与模板序列一致度极高,那么同源建模法是最准确的方法。如果目标序列与模板序列之间的一致度<30%,那么同源建模法是不适用的。
穿线法(I-TASSER)
原理:不相似的氨基酸序列也可以对应着相似的蛋白质结构。
我们可以把目标序列像线一样穿到目前现有的结构里,看穿到哪个结构里最舒服,哪个结构就可以作为预测的模板,并根据最舒服的穿法,构建出最终模型。通过能量方程可以知道穿的舒服不舒服。穿的舒服,能量就低,穿的不舒服,能量就高。穿线法就是通过计算目标序列穿到每一个已知结构中的每一种穿法下的能量,找到能量最低的那种穿法以及所穿的结构,然后把目标序列中的氨基酸替换到模板结构里来构建结构模型。
从头计算法(QUARK)
原理:1973年《科学》Anfinsen:蛋白质的三维结构决定于自身的氨基酸序列,并且处于最低自由能状态。
综合法(ROBETTA)
原理:综合了同源建模法、穿线法和从头计算法等多种方法,将氨基酸序列分段,情况不同的片段采用不同的方法。
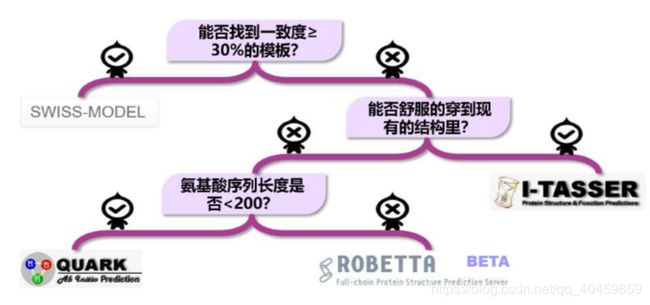 图 3 什么情况下用什么软件
图 3 什么情况下用什么软件
模型质量评估
模型质量评估软件(Model Quality Assessment programs,MQAPs)
对于通过计算方法获得的模型,必须进行必要的模型质量评估,以确定模型的可靠性。模型质量评估软件并不比较预测模型跟真实结构的差别大小,而是从空间几何学,立体化学和能量分布三个方面评估一个模型的自身合理性。
三级结构的比对
结构比对就是对蛋白质三维空间结构的相似性进行比较,它是蛋白质结构分析的重要手段之一。
- 可用于探索蛋白质进化及同源关系
- 改进序列比对的精度
- 改进蛋白质结构预测工具
- 为蛋白质结构分类提供依据
- 帮助了解蛋白质的功能
结构比对的结果可以用很多种参数来衡量,最常用的是RMSD。如果两个结构的RMSD为0埃,那么它们结构一致,可以完全重合;一般来说,RMSD小于3埃时,认为两个结构相似。
SuperPose 是一款在线蛋白质结构叠合软件。
SPDBV是一款蛋白质结构分析软件,也是一个蛋白质同源建模平台。其结构叠合功能十分出色。可进行整体只能叠合,或者选择性叠合。下载后无需安装,直接运行。
蛋白质分子表面性质
- 表面形状(VMD:SURF representation)
- 表面电荷分布(electrostatic potential)
- 表面残基可溶性(solvent accessibility)即残基与溶剂接触的程度,也就是哪些地方是掩埋在内部的(buried);哪些地方是露在表面的(exposed);哪些地方介于掩埋与暴露之间的中间状态(intermediate)。
蛋白质四级结构
蛋白质四级结构是独立的三级结构单元聚集形成的复合物,其中每个独立的三级结构称为亚基,也成为单体。含两个亚基的蛋白质称为二聚体;含三个亚基则称为三聚体;还有四聚体;五聚体;六聚体等。
 图 4 多聚体结构
图 4 多聚体结构
分子对接(docking)
蛋白质-蛋白质分子对接
对接过程中会考虑如下因素:
- 形状互补
- 亲疏水性
- 表面电荷分布
小分子化合物-蛋白质分子对接
刚性对接(Rigid Docking)
小分子总是柔性的,蛋白质上结合小分子的部分被认为是刚性的。
柔性对接(Flexible Docking)
小分子总是柔性的,蛋白质上结合小分子的部位被认为是柔性的。
虚拟筛选(Virtual screening)
虚拟筛选,也称计算机筛选,即在进行生物活性筛选之前,在计算机上对化合物分子进行预筛选,通过把一个蛋白质与多个小分子进行分子对接,以降低实际筛选化合物的数目,同时提高先导化合物的发现效率。
反向对接(Target Fishing)
通过把一个小分子与多个靶标蛋白进行分子对接,寻找潜在的靶标。
分子动力学模拟(Molecular Dynamin Simulation,MDS)
用计算机来模拟原子及分子的物理运动过程。