Nature综述 | 种内多样性:解释微生物组中的菌株

原文信息

题目:Diversity within species: interpreting strains in microbiomes
期刊:Nature Reviews Microbiology [IF: 34.208]
发表时间:2020.06
第一作者:Van Rossum Thea
通讯作者:Bork Peer
第一单位:European Molecular Biology Laboratory
原文链接:
https://doi.org/10.1038/s41579-020-0368-1
编译:范秋萍 云南大学 国际河流与生态安全研究院
摘要
传统上,研究种内变异仅限于可培养细菌的纯培养菌株和低分辨率微生物群落指纹图谱分析。宏基因组测序和其它技术的进步使在高通量和复杂环境中研究未培养微生物、高分辨率菌株和亚种成为可能。宏基因组分析具有巨大的科学前景,但也产生了大量描述亚种变异的方法和术语。本综述通过关注微生物学背景下的细菌和古细菌种内多样性来阐明这些进展。我们涵盖了与种群遗传学相关的基础微观进化概念,并概述了如何在微生物群落内直接研究和对种内变异进行分级,并重点关注宏基因组。最后,我们描述了如何使用宏基因组数据来实现物种内部变异的常见应用。我们旨在指导选择合适的术语和分析方法使研究人员从越来越高分辨率的微生物基因测序数据的获取中受益。
前言
一个多世纪以来,细菌培养已使数千种细菌菌株得以分离和分类。物种在细菌语境中被翻译为一群形成一致基因组簇的个体(有关详细信息和分歧,请参见下文)。尽管具有这种遗传相似性,但可以确定同一物种的菌株(同种菌株)之间可能存在很大的表型变异。在致病性的背景下,对种内变异的重要性进行了深入的研究,发现许多物种同时具有致病性和共生性菌株(例如大肠杆菌和Bacteroides fragilis)。确实,有研究发现大肠杆菌菌株可以是致病的、共生的、与宿主或与环境相关的。菌株特性与宿主健康之间的关系表明在物种水平上对微生物群落的研究可能是不充分的,并且在药物反应、养分循环、固氮和寄主关联等许多其它领域也有相同的应用。
在研究种内变异方面时基于培养的方法具有根本性和持续性的作用,尽管最近它们在方法上取得了进展,但仍具有较大的局限性。很少有微生物可以很容易地在分离的、实验室条件下获得培养,现有纯培养微生物是在典型的低通量条件下培养出来。即使可以进行培养,对微生物进行研究也是在分离后进行,而不是在其自然群落中进行。对整个微生物群落的非培养型菌株水平的分析已经进行了15年以上,但是受到测序深度浅和样本量较少的限制。随着宏基因组的技术和算法的创新(框1)以及测序成本的下降,有望对物种内变异进行大规模的宏基因组分析(框2)。这些方法有很大的前景,但同时也带来了科学和语义上的挑战。
在传统的培养方法中,“菌株”是指纯培养株或分离株,表示的是分类实体而不是自然概念,该操作定义不能直接转移到现代非培养的方法中。在高分辨率微生物组研究中出现了大量重叠的术语,这些术语通常定义不清。由此产生的混乱阻碍了微生物组领域和其它领域研究人员的交流和合作。为了在现有种内变异的定义下正确的纳入新的操作定义,必须了解创造和限制种内变异的微观进化过程。
在这项综述中,我们总结了产生和限制种内变异过程的同时描述了这些力量的平衡是如何塑造变异的大小和结构的。我们使用宏基因组数据概述了可用于研究这种变异并将其分级的主要方法,并在应用的背景下定义了常用术语。我们使用“种内变体”来指代物种水平以下的任何分组。在这篇综述中,我们重点介绍了使用宏基因组数据研究种内多样性的进步和挑战。
![]()
框1. 描述物种内变异的分子方法
在微生物群落中有很多种方法可用于研究种内变异。基于微生物组的方法较少,但不限于可培养的微生物群。诸如DGGE、TRFLP和ARISA之类的基础群落指纹识别方法使某些物种无需培养即可进行高分辨率研究。由于它们的通量和分辨率低,这些方法在很大程度上已被基因测序方法所取代。尽管16S rRNA基因扩增子分析起源于低分辨率方法,但现在有时可以在整个基因序列中使用Oligotyping、扩增子序列变异体(ASV)和单核苷酸变体区分某些物种。然而,在种内分析的分辨率上,16S rRNA的方法仍然受到极大限制,可能被每个基因组中16S rRNA基因的多个非相同拷贝所混淆。
鸟枪法宏基因测序为标记基因或者全基因组提供了更多的信息。已经开发出许多工具分析宏基因组数据以描述物种内变异。主要方法包括在预定义的标记基因内还是在整个物种参考基因组,单核苷酸变体与参考菌株基因组的总体相似性以及序列分类和基于基因含量的结构分析。基因组组装的基因组可以通过装箱和组装共同丰富的基因来恢复。然而,这些都有重要的局限性(框3)。
基于微流控的技术可在测序和单细胞测序之前富集特定生物体,从而产生单个扩增的基因组。由于方法学的进步,我们能培养越来越多的细菌,该技术结合了多种培养条件和细菌的快速鉴定。
基于冷冻电子显微镜的成像和转录组以及基于蛋白质组和代谢组学的分析方法都是非基因组方法,可以捕获物种内的表型差异,可以单独使用,也可以结合基因组方法使用。
![]()
![]()
框3. 在宏基因组中研究物种内变异的挑战
对微生物群落种内变异的调查面临研究设计、技术和方法上的挑战。研究设计的主要挑战是“无法观察到的变化”:您看不到未采样的内容。如果一个物种内变异性低,这很难证明这不是由于采样欠缺或采样偏差引起的。这种偏差可以是暂时的(例如,由于菌株转换或灭绝)或空间的(例如,由于邻近采样区域隐藏着很多不同的同物种结构,例如土壤或皮肤)。较浅的测序深度也不利于观察物种内部低丰度的变体。越来越多的深度测序宏基因组样本减轻了这些偏差,但这些样品在整个研究中的整合仍然面临宏基因组众所周知的一些技术挑战。
宏基因组组装基因组(MAG)对于研究未知和代表性不足的物种是很有用的,但也具有重要的局限性。MAG是种群共有基因组。因此,基因座位可能是未连接的多等位基因。与分离的基因组相比,MAG的装配质量通常较低、不完全并且更可能是嵌合的。由于难以检测低于物种水平的嵌合体,因此不应将MAG等同于分离株的基因组测序。与完整分离株基因组相似的MAG才能使用“完整MAG”一词,该基因组通常是没有间隙的单个圆形拼接体。
为避免将分离株基因组和MAG混淆,应避免将MAG上传到公共基因组数据库的做法,并且不应将“基因组解析的宏基因组”这一短语用于不直接评估MAG内异质性的MAG研究。单细胞测序方法为从宏基因组中回收基因组提供了一种替代MAG的方法,但单分子DNA的使用受到高成本、低通量、潜在污染和质量问题的限制。
持续的技术进步,测序成本的降低以及互补方法的日益集成对于应对数据生成和集成中的这些挑战将是必要的。
![]()
物种内的变异和内聚力
导致种内变异的过程
种内多样性是由变异和随后的选择和漂变造成的(图1)。突变和基因流使遗传变异进入其它相同的克隆子细胞谱系中。
DNA复制过程中的错误、DNA修复和重组机制的错误使基因组中不断出现突变(即替换、插入、缺失和反转)。尽管在双螺旋DNA中109个核苷酸复制一次才约有一个核苷酸改变,但物种间和物种内部的突变率可以相差一个数量级。选择较低或更高的比率可以平衡突变频率降低的代谢成本以及有害突变的影响。这种平衡的方向取决于栖息地条件、种群大小和等位基因突变强度。一个细菌谱系内突变的累积速率取决于突变率以及影响突变的自然选择和遗传漂变。此外,并非所有的细菌基因组都容易发生突变,除非核心基因位于辅助基因或可移动遗传基因附近,辅助基因的突变累积率才高于核心基因,次级染色体中的突变累积率高于初级染色体。一般来说,缺失比插入更为频繁,非功能性序列很容易从细菌基因组中丢失。一个基因组中发生的突变可以垂直传递给后代,也可以水平传递给邻近的细胞。
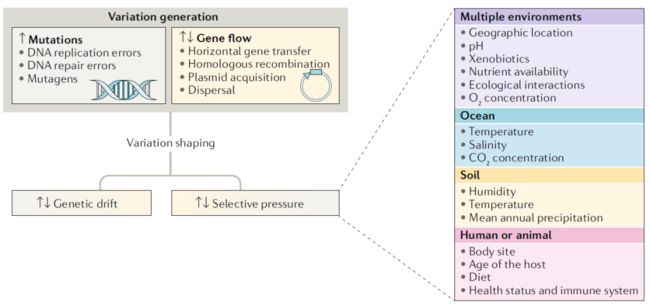
图1. 细菌种内变异的主要驱动因子
从一个物种转移到另一物种(基因流)的遗传变异会使基因组重排。DNA可以通过转化、转导、接合、转染和膜囊泡等水平基因转移(HGT)途径在细胞之间转移DNA。新获得的供体DNA可以作为质粒或裂解噬菌体在受体细胞内保持分离,或者可以通过同源重组等多种机制掺入受体基因组中。HGT在种内更常见但也在种间也存在。HGT可以通过物种内同源重组用供体同源物替代遗传片段,获得新的遗传物质。就HGT对物种内变异的影响而言,最重要的不是同源重组而是所传递的遗传物质对受体物种而言是否是新的(如下所述)。结合过程缺乏表面相容性、CRISPR介导的微生物免疫力和噬菌体宿主特异性受限是限制HGT的主要过程。
自然选择和遗传漂变决定了通过突变和基因流引入的物种内部变异。遗传漂变随机消除了种群内的遗传变异,而自然选择则维持或消除了带来适应性优势或劣势的变异。自然选择是由多种生物和非生物因素驱动的,这些因素不同程度地影响了亚群生存和复制的能力(图1)。这些因素可以通过群落聚集和经典进化力来影响物种间和物种内微生物群落的组成。选择压力因子随生境而异,可能包括pH、温度、氧气和其它气体的浓度、养分的可利用性、与其它细菌的直接竞争或合作、噬菌体和真核生物的捕食以及压力诱导产生的药物、抗菌化合物和重金属等异源生物的存在。
物种定义和物种内聚力机制
通过突变的垂直积累和基因的水平获取,一个细胞后代之间的变异可能会不断增加,从而在整个细菌基因组中形成连续的遗传变异景观。但是,当比较细菌之间的基因组相似性时,会观察到不同的簇。尽管人们质疑“物种”概念的适用性,但这些簇被认为是细菌中的物种。在本综述中,我们使用“物种”一词来反映这些遗传相似性簇。
数十年来,已经使用DNA-DNA杂交(DDH)来测量基于基因组相似性的细菌种类。根据细菌命名法,通过DDH,同种基因组具有≥70%的相似性。DDH逐渐由分离株的DNA测序和平均核苷酸一致性(ANI,average nucleotide identity)比较来补充或取代,DDH相似性≥70%对应于核心基因组中ANI≥94%和通用标记基因中ANI≥96%。这些对应关系中的近似值可能会影响分类,如通过DDH对核梭菌(Fusobacter nucleatum)进行分类后建议通过ANI将其重新分类为单独的物种。早期研表究表明使用宏基因组数据可以识别出独特的细菌物种边界。最近,通过研究基于整个基因组(~95%)和标记基因(96.5%)在ANI阈值处确定该边界并描述了核心基因组中的基因流急剧下降。
尽管ANI基因组数据具有总体一致性,但对细菌和古细菌物种的定义仍存在争议,关于“物种”的定义有20多个,并对此概念存在质疑。物种的生物学和系统发育概念最适用于细菌和古细菌。前者将物种定义为一组可以通过杂交产生有活力后代的个体,这意味着原核生物可能会发生同源重组,而后者则将物种定义为具有独特表型特征的进化枝。这两个概念都预测不同物种之间的同源重组速率和HGT速率会下降。仅基于基因组的ANI比较并不一定能很好地满足对众多潜在物种的定义,相反可以使用其它方法来定义除了ANI之外的物种或替代ANI的物种,例如通过表型、通用单拷贝基因(例如16S rRNA基因)中的相似性和基因含量来定义物种。
物种内的基因组相似性称为“内聚力(cohesion)”。这主要通过种内重组和选择低适应性等位基因来维持。如果等位基因比某个群体中的其它所有基因更有利,那么它可以在该群体中完全传播产生硬性选择性扫描。当重组率较低时,整个基因组可能会借助适应性等位基因使其流行,从而产生全基因组选择性扫描。当发生硬性全基因组选择性扫描时可以减少物种内多样性,保持物种间相似性。
决定物种内部变异大小和结构的因素
物种内多样性产生、维持和清除的程度不同,因此某些物种具有高度异质性,而另一些物种则具有紧密的内聚力。种内变异的这些特征取决于所观察到的种群(框3),并可以整体或局部描述。增加多样性与保持内聚力之间力量的平衡决定了物种内变异的程度和结构。
物种的变异量取决于突变率、产生时间、种间HGT的倾向和种群大小,而持续存在的变异量取决于生境中选择压力的严格性、种群大小以及选择性扫描的频率和程度。差异和内聚力之间的平衡是由选择和漂变调节的,而选择和漂变是由生物和非生物因素决定的(图1)。如果所转移物质对受体种群而言是新的,那么HGT可以增加种群内的遗传变异,例如,如果供体细胞是从外来种群中散布的或远缘相关的,那么种内遗传变异增加;相反,如果HGT将遗传物质散布到整个种群中,则可以根据特定基因含量或单核苷酸变异(SNV)使种群均质化,从而产生基因特异性的硬性选择性扫描。
在一个物种内,结构化群落可以通过软性选择扫描的组合(当多个替代适应性等位基因在种群中扩散并共存时)使其沿着漂变和扩散到相似的或新的生态位。例如,当突变率高而种内重组的速率低时,菌株可能会分化成更具内聚力的亚组。具体而言,重组与突变之比在0.25以下时可以使亚种群自由扩散,这可能使能够在物种形成过程中破坏部分基因流的亚种形成。
亚种可能是由物理或地理障碍引起或加速的,这些障碍阻碍了亚种之间的基因流动(异源),自然选择或漂变使亚种产生差异。但是,在没有空间隔离的情况下也可以产生亚种。在这种情况下,专业化可能具有选择优势,可以减少对资源的竞争。由于细菌和古细菌的扩散能力极高,对基因流来说,完整的物理阻隔可能很少,两种情况都有可能发生。当出现偶然的基因流动以及生态位重叠时,对选择进行纯化可以维持亚种之间的部分内聚力,这可以避免建立稳定亚种时产生差异。
在一种极端情况下,物种可能是单型的,也就是说,它们在整个种群中的遗传相似性分布均匀或“模糊”。多样性较低的单型物种更可能是专一的,地理分布或寄主范围狭窄,或者是最相近形成物种的产物。在另一种极端条件下,具有亚种(多型)和高度多样性的物种更可能是自由存在的通才,能适应不断变化的环境,具有广泛的地理范围或许多部分重叠的生态位。
上述许多知识都是通过培养和分离实验逐个物种获得的。微生物学方法的兴起使得我们能够大规模地表征物种之间的变异,并新的提供可行的研究途径(框2)。将这些发现有意义地应用于上下文中对于适应这一知识体系中的概念和术语并用于宏基因组研究非常重要。
![]()
框2分离株培养与宏基因组分析物种内变异的比较
传统上,对物种水平以下进行研究依赖于培养的分离株。随着宏基因组的兴起,高分辨率遗传数据增加了。通常,这些数据是基于特定遗传片段(例如标记基因)内的变异或通过组装回收的基因组(由基因组组装的基因组;MAG)中的变异进行分析的(框1)。尽管生成的数据量巨大使空前的发现成为可能,但这些新方法也具有重要的局限性并带来新的复杂性(请参见下表)。尽管宏基因组比研究分离株更有优势,但这两种方法仍是互补的。为了确保两种方法之间的协同作用,分离基因组和MAG数据质量必须易于获得且具有可比性,并且应维持通用词汇表。
![]()
基于分离培养和宏基因组测序的种内变异研究

种内变异的分级
通常需要将物种内变异分为有意义的组进行研究,并与诸如健康状况、地理位置或代谢能力等分类变量相关联。上面描述的理论可以支持此类群的概念定义,但通常不能直接在微生物研究中使用。相反,必须基于可衡量的标准来设计变体群组的操作定义。通常,这是在遗传或表型尺度上完成的。在操作上定义变体群组(例如菌株)的适当度量取决于所问的生物学问题和所使用的方法(图2a)。
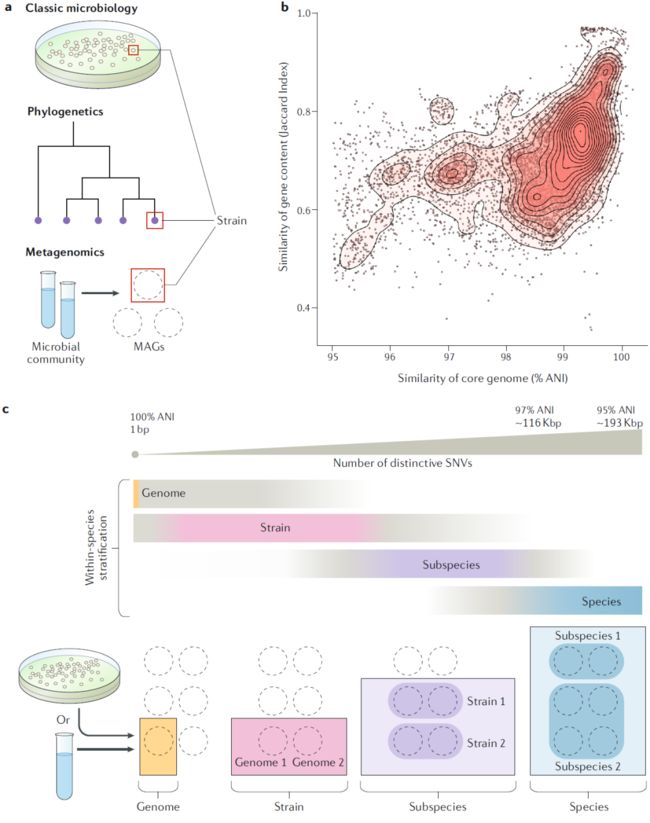
图2. 种内变异的分级。a:基于研究领域“菌株”的各种可操作分类定义;b:每一个点都是一个分离株基因组与所有其它同种分离株基因组的成对比较。
使用宏基因组数据进行遗传分级
物种内的遗传变异可以通过多种方式来衡量,其中一些共同的指标是总体基因组相似性以及共有基因和独特基因的数量和/或SNV的数量和性质。在本节中,我们讨论如何采取这些措施,并探讨其优势和局限性。当将这些分析方法应用于宏基因组测序产生的大量数据时可以以高通量方式同时对多个物种进行物种内结构分析,但这也引起了诸如数据不完整和数据存在部分错误的各种数据质量问题,也存在大的计算需求和存储需求等技术挑战。
同源基因组在亚种水平上的总体相似性可以通过序列数和参考基因组中直接获得的宏基因组数据进行评估或者通过比较宏基因组聚集的基因组(MAG)来评估。合适参考基因组的可用性低会限制基于参考基因组的方法的使用,尤其是在非人类微生物组中。现在可以使用大量的MAG,并且计算ANI方法的效率也得到了提高。但是,对于大型基因组序列ANI的计算仍然具有挑战性。此外,由于数据质量的局限性和不完整性,在ANI比较中使用MAG可能会引入不准确性(框3)。
ANI和重组速率下降表明正在对一个物种进行细分。但是,与物种边界相反,物种内变种没有显示出基于基因组或标记基因的通用阈值,基因组或者标记基因能够将种分为几组。相反,物种内ANI值的分布和范围因类群和种群而有所不同,这限制了其在分级中的效用。此外,由少量核苷酸编码的遗传差异对表型的影响很大但对ANI的影响很小。因此,在物种内ANI差异较小的情况下,基因含量、SNV和indel的测量值在定义生物学上相关的物种内变体方面提供的信息比ANI多。
基因含量是基因组中所有基因的总和,包括几乎存在于所有亚种变体中的核心基因和仅存在于一个亚种中的辅助基因。变体之间辅助基因含量的差异可能在单基因水平或遗传片段水平出现,其中可能包括多个基因(结构变异),可以根据是否存在基因或该基因的拷贝数来计算基因含量差异。认为在结构变异内存在基因顺序,但尚未直接通过宏基因组方法解决。通过寻找基因簇或通过将基因含量与SNV结构定义的变体相关联,宏基因组数据可用于研究物种内基因含量的变异。HGT使基因含量相似性和系统发育之间的关系复杂化。但是,对同种基因组的比较研究表明基于基因含量的配对相似性与基于核心基因组ANI的成对相似性相关(图2b),并且不同的SNV结构可以对应于不同的基因结构。
SNV差异可用于比较高分辨率的同种变体。这些比较可以考虑变体位置的数量,在核心基因、辅助基因或基因间区域中的位置,它们在基因组中的传播(聚集或扩散)及其潜在的表型影响(同义或非同义突变)。在宏基因组中,SNV的鉴定可以基于MAG或从现有的参考基因或基因组。参考的群落成员与实际群落成员之间的相似程度可能会对结果的准确性产生重大影响。基于MAG识别的SNV可以揭示种群动态,但是由于MAG参考的基因潜在质量低,也可能产生错误(框3)。可以基于不同的SNV(例如SNP类型)从宏基因组数据中定义同种基因组群组。用于显示单个SNV差异的分离株数据可以确定致病性或抗菌药物耐药性。当测序深度较浅且种群规模较大时,不利于丰度较低的SNV的检测。当SNV垂直转移时可用于定义单倍型和谱系。可以进一步扩展这种方法,将SNV用于重建物种内的系统发育,但必须注意使用不太可能位于HGT区域的基因座。
当多个遗传变异出现在一条染色体上时,它们是“连着”的。连着的变异可以一起遗传,但可以被重组或突变破坏。确定等位基因之间的联系可以用来追踪谱系、重建单倍体类型(阶段性变体)以及检测HGT。然而,当使用典型的短片段、鸟枪式测序方法时,在提供联系数据方面,宏基因组数据存在固有的局限性,因为这种方法会分解DNA。短序列的组合可能能够记录联系的基因信息,但当一个样本中存在多个高度相似的基因组时会产生很多嵌合体,例如多个同源菌株(菌株异质性)。鸟枪宏基因组通常不提供联系等位基因的精确图谱,而仅限于提供一组具有等位基因频率信息的多等位基因座;如本综述最后一节所述,这些方法仍然可以用于许多应用,还可以用其对一个物种进行种群遗传分析,例如计算种群多样性、种群结构和选择压力。
已经开发了许多软件工具以使用宏基因组数据对物种内的多样性进行测量和分类。通常,它们有分类和发现这两大目标。面向分类的工具有metaMLST、PathoScope、MetaPhlAn2、StrainSifter、Sigma、SPARSE和StrainEst,目的是检测样品中是否存在已知的、特征化的物种内群组。面向发现的工具通常使用以下三种方法中的一种将物种内部变异按相似性簇进行分组:基因含量(例如PanPhlAn),整个或核心基因组中的SNV(例如metaSNV)或标记基因中的SNV(例如,谱系算法、ConStrains、StrainPhlAn、DESMAN、StrainFinder和mOTUs2),然后检测独特的基因内容(例如DESMAN)。尽管许多工具声称可以提供菌株水平的分辨率,但术语“菌株”在软件中的定义有所不同(有关定义的讨论,请参阅下一部分)。可以从整个样本的SNV多度中恢复SNV联系信息的工具包括ConStrains、DESMAN、StrainFinder和Lineages算法。当假设样本包含物种内单一优势种群时,也可以用StrainPhlAn和metaSNV之类的工具将SNV聚类为物种内群组(分别为菌株和亚种)。
尽管这些工具支持用宏基因组数据来研究物种内变异(请参见下文),但它们具有一些较大的局限性。例如,依赖于绘制参考基因组或标记基因序列图的工具固有地受到适宜参考基因组可用性的限制,在某些环境下(例如淡水和土壤),参考基因组的利用率非常低。可以通过构建和使用MAG来规避此限制(例如,在DESMAN中),但是必须考虑MAG质量,特别是在没有时间序列数据的情况下(框3)。其它限制包括极高的覆盖深度和无法处理大量数据。这说明随着宏基因组领域向更大、复杂的数据集发展的过程基础软件的局限性是如何产生的。这些局限性和其它局限性结果导致某些工具难以运行或无法运行,或者无法与当前大小合理的数据集一起使用,从而无法再现或扩展结果。
本综述参考的软件可以执行所描述方法论方法中的工具示例。这些参考不是对准确性或可用性的认可或报告。最近的综述中对许多工具的功能进行了比较,尽管预期在宏基因组解释框架的关键评估(CAMI)中会解决这些问题,但尚未完成对准确性的彻底比较。预计未来的工作将对种内分析软件进行比较;但是,每种工具特定术语(例如,SNV类型,菌株种群等)的确切含义以及它们对通用术语(例如,菌株和亚种)的映射都必须谨慎处理。
基因分级的术语
有许多术语可以对物种内的变异进行分级(表1)。从国际原核生物命名法中最常用和公认的术语中我们重点对基因组、菌株和亚种这三个涵盖物种内遗传变异范围的术语进行介绍(图2c)。在本节中,我们讨论在基于培养的微生物学和宏基因组中使用这些术语时的冲突,并提出解决方案。
几十年来,微生物基因组最常见的是来源于分离株的测序。近来,MAG的快速产生,以及超越了分离株基因组的应用。分离株和宏基因组之间协同作用的障碍是将MAG等同于分离株基因组(框3)。前者可能代表包含有多样性非常多的种群,而后者通常代表了多样性几乎没有的培养分离株。考虑到单细胞测序的兴起,将“基因组”一词视为细胞、分离株或宏基因组是有用的。
“菌株”一词广泛应用于微生物学的各个领域,且有许多不同的定义(图2a)。在细菌学中其定义是:一个在纯培养条件下产生的单一分离株后代组成的,通常是由在一个或多个细胞中发现最初的单个菌落,最终衍生出来的一系列培养物组成。在这种情况下,菌株是起源于分离的。在流行病学中对菌株有另外的定义,认为菌株是作为子实体存在于自然界中的。这种“自然菌株”被定义为一组具有独特基因型和/或表型特征的同种分离株。可以将“分类菌株”视为自然菌株的分离培养物。在操作上,自然菌株和分类菌株的边界各不相同。例如,分类菌株可以表型异质,即使具有至少三个突变,也被称为同一菌株。相比之下,在某些情况下,分离株需要少于三个SNV差异才能被认为来自同一自然株。这表明在以培养为核心的微生物学中尚未普遍设定菌株描述的遗传阈值。
表1. 描述种内变异的常用术语

在以培养为核心的微生物学中,菌株的这两个定义继续共存,在微生物学中采用这一术语扩大了这种复杂性。很少使用前缀带有歧义的“分类”和“自然”。但是,这种二元性可以阐明“菌株”一词在宏基因组中的混合用法。菌株水平的宏基因组经常提出分类和发现这两种类型的问题。分类问题会问遗传片段(测序片段数)是否属于特定的分类菌株,例如检测粪便样本中是否存在益生菌菌株双歧杆菌BB12。发现问题会问物种内是否存在形成自然菌株的亚组,例如通过聚类基因组或遗传片段的遗传变异。用于菌株发现的宏基因组工具之间可能会产生冲突,这些工具使用不同的自然菌株定义,因此会给出不同的结果,例如,基于差异基因含量和共享基因中的SNV来定义自然菌株。
尚未对菌株建立一个能够普遍适用,具有牢固生物学基础的操作定义,这也可能是不存在的。从理论上讲,可以将只有一个SNV差异的基因组称为不同菌株。但是,由于宏基因组数据能产生大量的菌株,因此不建议使用此方法。对于多少个SNV定义一个分离菌株以及是否需要在种群中固定这些SNV或者需要影响其表型没有规定。在实践中,在菌株水平工具选择中如何选择设置此临界值(例如,在StrainPhlAn中设置物种特异性标记基因的核苷酸超过0.1%)或者由分析作者设定的临界值是不确定的(例如,ANI大于98%)。鉴于菌株的操作定义具有这种可变性,因此使用更具体的术语代替“菌株”的通用术语变得特别有价值(有关指导,请参见表1和标题为“种内变异的应用”部分)。
亚种是群组的同种菌株,该术语存在许多定义。在经典微生物学中,亚种是遗传或表型不同的菌株簇,具有可用的类型菌株并被命名(例如,枯草芽孢杆菌亚种)。随着时间的流逝,亚种分类的基础已经从定性表型方法转变为分离株之间的基因组相似性。这种变化导致了分类转换,例如物种降级为亚种(例如,在长双歧杆菌中),反之亦然(例如,在必要的多核细菌中)。因此,已命名的经典亚种不一定与不同的基因组簇对齐。相比之下,在种群生物学的背景下,亚种是一组生活在一个物种空间范围内的局部物种,并且与相同物种的其它种群在基因型或表型上不同。将“亚种”一词用于微生物组意味着这与菌株描述的二分法用法相同,即将序列分类为现有的“经典亚种”和“种群亚种”,种群亚种是通过空间尺度上的种内遗传变异进行聚类的。
尽管这些术语的严格定义并不限制它们各自可以包含的相对变化量,但实际上,将它们放在彼此的背景中并在建议的范围内使用它们是有用的(图2c)。由于这些范围是准则,因此在使用每个术语时应在报告中包括群组划定的实际阈值。重要的是,“菌株”从属于“亚种”,因此不应被普遍用来指任何从属于物种的分组(有时是这样)。由于定义不同,但在视觉上与“亚种”相似,因此我们也不鼓励用“亚种”一词来代替“物种以下”。相反,我们建议使用“出现在同种中的”或“种内”一词。例如,“菌株水平分析”或“亚种分析”的不当用法将由“同种物种分析”或“种内分析”代替。此外,物种内的非特定群组也可以称为“物种内变体”。
微生物群落的表型分级
一个物种内的遗传变异可以以复杂的方式表现为表型差异。不同的遗传变体可以表现为相同的表型,而相同的遗传变体可以在不同条件下表现为不同的表型。遗传差异的大小及其表型影响不一定相互关联,例如在抗生素耐药性急剧增加的情况下,只需少一个SNV即可。此外,当细菌在分离培养或共培养或在其自然群落中培养时,可以观察到不同的表型。例如,铜绿假单胞菌在体外和人类感染期间有不同的基因表达结构,包括与抗生素抗性、细胞间通讯和代谢相关的基因,对治疗的发展具有影响。在物种内部也可以看到表型的差异,例如两株嗜盐细菌盐沼细菌的菌株在分离培养时具有相似的表达模式,而在共培养时则具有不同的表达模式。这些例子强调了直接在微生物群落中研究物种表型变异的重要性,这可以通过几种方法来完成(框1);例如,超转录组学已被用于揭示贻贝中同种共生体之间的功能多样性,并且通过宏基因组学推断的复制率已经区分了婴儿中Citrobacter koseri 的亚群。
基因型和表型之间的复杂关系意味着表型分类方案可能与遗传分级不一致。在医学和流行病学中,基于不同的致病性(病理型)或细胞表面抗原(血清型)将细菌分类为群组(可能是多系统的)是有用的。例如,肠道大肠杆菌群包括共生菌株和致病菌株,它们被分为至少七个致病型。在生态学中,还可以基于行为及其在群落中的功能角色来定义群组,例如,根据所使用的资源类型和开发方式来定义群组。以这种方式分组的物种称为“公会”,这一概念和术语可以类似地用于描述菌株群组。此类分组旨在为生态系统内的竞争分析提供适当的解决方案并在整个群落中推广。尽管表型与许多生物学问题最相关,但很难进行大规模测量。通过微生物基因组测序,基因型在高通量条件下更容易测量,但是将它们与表型联系起来具有挑战性,因为表型会随生境和较小的基因型差异而急剧变化。
种内变异的应用
种内变异的多种尺度和维度反映了物种内调查可以解决许多生物学问题。基于分离的方法已用于调查涉及种内变异的许多生物学问题。随着宏基因组方法的兴起,现在可以在高通量下调查一些相同的问题,也可以同时对群落中的许多物种进行研究(有重要限制;框2;框3)。下面,我们将描述基于分离方法开创的重要的生物学应用现在有多少可以使用宏基因组方法进行研究。我们将这类调查的常见例子归纳为5大主题,围绕关键生物学问题开展(图3)。对于每个主题,我们总结了方法论方法和适当的术语,并提供了相关研究或软件的示例。
来源追踪
此样本中的细胞最初来自哪里?为了确定微生物细胞传播或扩散方式,必须确定其确切来源。通过比较来自目标细胞或种群的遗传物质与其潜在来源种群或祖先的遗传物质进行比较,可以计算出细胞从特定来源种群中扩散或成为特定来源种群直接后代的可能性(来源追踪、传播追踪或谱系追踪;图3a)。从宏基因组数据确定源种群的策略包括检测共享SNV、CRISPR信号或菌株特异性基因的存在以及基因组重建。这些方法已用于评估是否有细菌细胞从人的口腔传播到肠道,从母亲传播到婴儿,从益生菌治疗传播到消费者或从粪便微生物组的捐赠者传播到受体。这些策略可能被等位基因连接的宏基因组破坏,从而使多个来源种群以及目标种群的进化变得复杂。因此,尽管谱系追踪方法可用于病原体源检测,但对于流行病爆发的分析可能不够。在源追踪中,可以用更具体的术语“谱系”来代替通用术语“菌株”,该术语可以用单倍型来表征。由宏基因组数据确定基因组单倍型仍然是一个挑战。然而,随着错误率的降低,有望实现单个DNA分子的长序列测序。
系统发育重建
该物种内中变体的进化史是什么?在系统发育重建中,从遗传相似性推断一个物种内多个谱系的相关祖先(图3b)。这种相似性可以基于完整的基因组或遗传片段(例如标记基因)获得。由于HGT和同源重组,重建的系统发育可能会根据所选择的基因座而有所不同,可能无法用遗传片段的系统发育反映总基因组的系统发育。可选择地,物种内的系统发育研究可能集中于重建物种内特定基因的历史或质粒的历史。物种内系统发育把这些历史放在观察到的地理分布背景下。使用分离基因组进行系统发育分析已得到很好的实践,如果回收了高质量的基因组,例如使用MAG或单个扩增的基因组,这些方法可以应用于微生物群落。但是,在此应用之前必须考虑数据质量问题(框3)。另外,一种典型的方法是在宏基因组中鉴定同种、同源基因片段(例如,通过比对参考序列),检测其中的SNV,然后推断其最可能的历史。可以基于系统发育来定义物种内的群组,方法是在任意相似度上将生成的树切割为任意相似度,从而创建“系统型”。在这种情况下,可以用更具体的术语“ 进化枝”或“系统型”代替通用词“菌株”。
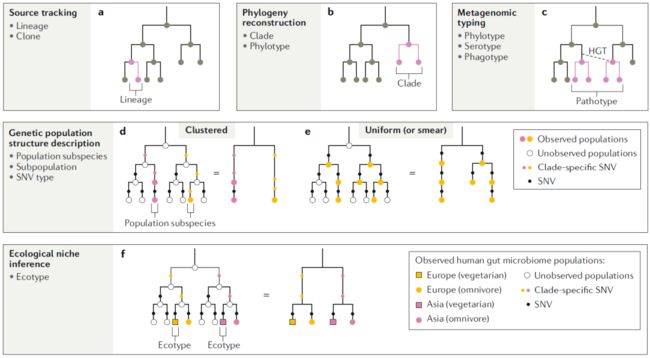
图3. 种内变异的应用
遗传种群结构描述
该物种是否具有不同的亚种群?描述物种的遗传种群结构可以表明其地理历史或解释与宿主疾病状态的异质性联系。物种的种群结构可以通过与观察数据重叠的遗传数据确定以描述种内和种间变体遗传相似性的分布。当在所观察到的物种变体中遗传相似性具有平滑分布时,就会出现均匀的结构(涂片)。当祖先和姐妹进化枝种群存在时,即在树内几乎没有未观察到的种群不时,就会发生这种情况(图3e)。相反,当遗传相似性之间存在不连续性时,会出现簇状结构,从而使进化枝可以分为不同的簇。这种不均匀的结构是由树内灭绝的分支所形成的(图3d)。这表现为亚群,即整个种群中遗传变异频率不同(例如,等位基因或SNV)的子集。
通过寻找潜在亚群之间遗传相似性的聚类,宏基因组可用于研究微生物群内的种群遗传学。检测亚种群对采样工作很敏感,因为遗传相似性的不连续性可能是因为未能观察到中间产物(框3),可以根据整个基因组中SNV等位基因频率,标记基因中SNV或基因含量差异来评估此类遗传相似性。当MAG或单个扩增基因组产生时可以使用基于基因组ANI进行聚类。MAG还可以用于追踪SNV和基因含量差异,在这种情况下,有时用“菌株”来指代一个或多个亚种是不恰当地。如果亚种适应了不同的生态位则可能是生态型,例如通过基因组广泛扫描而不是基因特异性扫描。
生态位推断
该物种内的变体是否适应了不同的条件?研究物种内变体及其生境可以提供生态位特异性的信息(图3f)。当使用遗传数据推断未描述的栖息地时,有时被称为“反向生态”。这些查询通常旨在确定那些对于适应特定环境至关重要的遗传片段(例如,基因、操纵子或质粒)。这些片段的获取可能来自垂直传播或水平传播,因此可能与该物种的系统发育史形成鲜明对比。例如,由于在选择性条件下(基因特异性扫描),HGT频繁发生,因此一个基因能够在种群间广泛传播,例如在抗生素存在的情况下,HGT频繁发生使得基因可以在人群中迅速普及。使用宏基因组数据研究这些问题的常用方法是查看已适应不同条件的同种亚群细胞(例如,不同人类宿主饮食、土壤与植物宿主或湖水栖息地的变化),然后确定独特的基因。基因组宽度联系研究中使用的方法也可以在这里应用,尽管这些方法通常不关注种群的适应性进化。在这种情况下,一般术语“菌株”可用更具体的术语“生态型”代替。
在图3所示的例子中,遗传种群结构调查将关注于欧洲和亚洲种群的等位基因频率差异,以决定这些种群是不同的亚群还是属于一个连续种群。无论欧洲和亚洲人口是否是不同的亚群,生态位推断研究将关注与不同饮食相关的肠道微生物组物种的基因差异。
分类
该物种变体是否属于先前描述的物种亚组?分类分析评估了同种物种变体中具有特定目的的遗传特征(例如SNV、基因、操纵子或质粒;图3c)。在这种情况下,物种内群体的确定不是基于进化史或栖息地范围,而是根据是否存在特定遗传特征。这样的特征可以赋予栖息地适应性,可以是短暂的,并且仅在抗微生物抗性基因、致病性基因(例如,肠致病性大肠杆菌)或鞭毛等罕见或人工条件下表达。在这种情况下,主要考虑HGT,因为其结果意味着遗传特征的存在并不一定反映系统发育。
宏基因组方法可用于检测定义分类的遗传特征。可以基于参考序列检测已知SNV或新SNV的重要性。在宏基因组中已经基于与参考序列的同源性检测搭到分类基因的存在,但是群落内可能存在HGT,因此很难确定这些基因是否存在于特定菌株中。在宏基因组数据中,无论有无组装基因组,均可直接研究HGT。
物种变体内相同表型的比较分析能发现与该表型相关(并可能引起该表型)的特定遗传特征(例如在基因组宽度相关的研究)。例如,仅基于它们在显示相似症状的宿主中存在就可以将同种细胞分组为致病性“变体”,而无需了解细胞或其典型栖息地的进化关系。在这种情况下,一般术语“菌株”可以用更具体的术语“病理型”代替。
传统上,使用分离基因组方法或低分辨率分子方法对上述主题进行了研究(框1)。随着宏基因组研究产生了大量数据,已经建立了数十种新方法来研究相同的问题,通常使用它们自己的新词汇。考虑如何将这些新方法映射到它们要解决的基本生物学问题上以及该领域的研究历史将如何控制术语的爆炸式增长。许多研究将上述主题组合在一起,但是单独考虑基本单元将有助于分解复杂问题并选择最合适的方法和术语。
结论
尽管在微生物组调查中,通常考虑的是最高分辨率的分类类别,但物种仍可能具有极端的表型变异性。过去使用的是一些基于分离方法获得的数量有限的可培养细菌,因此研究这种变异性的范围相对有限。随着宏基因组测序的发展,可以研究的物种数量和可以使用的方法数量大大增加。根据许多标准和尺度,对物种内部变异进行分级也使术语增长和不精确。揭示物种内部变异是如何产生的,并确定所要研究的核心生物学问题,有助于确定正确的术语和方法。在某些情况下,最合适的术语可能具有操作性定义,并且其详细信息和阈值可能因研究而异。为了促进沟通和协作,能对宏基因研究进行比较,应尽可能避免使用没有严格定义或广为人知的术语。本综述旨在指导此类描述,并支持将物种内调查技术更广泛地应用于宏基因组学数据的开发和应用。
引文:Thea Van Rossum, Pamela Ferretti, Oleksandr M. Maistrenko & Peer Bork. (2020). Diversity within species: interpreting strains in microbiomes. Nature Reviews Microbiology, doi: https://doi.org/10.1038/s41579-020-0368-1
猜你喜欢
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树
必备技能:提问 搜索 Endnote
文献阅读 热心肠 SemanticScholar Geenmedical
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
在线工具:16S预测培养基 生信绘图
科研经验:云笔记 云协作 公众号
编程模板: Shell R Perl
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。PI请明示身份,另有海内外微生物相关PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。

学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”
 点击阅读原文,跳转最新文章目录阅读
点击阅读原文,跳转最新文章目录阅读