建议查资料来源:
1、 微信搜索,很多公众号写的比较全
2、 CSDN代码解读比较好,相关小点也说的比较好。报错代码一部分也能查到。
3、 博客园
4、 简书
5、 谷歌
一、了解到底什么是WGCNA。
先通读了解相关概念。先不要去纠结代码。看最基础的概念就好,实在理解不了,那,那就算了叭,毕竟后面视频还是会讲的,逃不过的……但是WGCNA分析大概一个什么流程是的知道的。
加权基因共表达网络分析 (WGCNA, Weighted correlation network analysis)是用来描述不同样品之间基因关联模式的系统生物学方法,可以用来鉴定高度协同变化的基因集, 并根据基因集的内连性和基因集与表型之间的关联鉴定候补生物标记基因或治疗靶点。
相比于只关注差异表达的基因,WGCNA利用数千或近万个变化最大的基因或全部基因的信息识别感兴趣的基因集,并与表型进行显著性关联分析。一是充分利用了信息,二是把数千个基因与表型的关联转换为数个基因集与表型的关联,免去了多重假设检验校正的问题。
理解WGCNA,需要先理解下面几个术语和它们在WGCNA中的定义。
- 共表达网络:定义为加权基因网络。点代表基因,边代表基因表达相关性。加权是指对相关性值进行冥次运算(冥次的值也就是软阈值 (power, pickSoftThreshold这个函数所做的就是确定合适的power))。无向网络的边属性计算方式为
abs(cor(genex, geney)) ^ power;有向网络的边属性计算方式为(1+cor(genex, geney)/2) ^ power; sign hybrid的边属性计算方式为cor(genex, geney)^power if cor>0 else 0。这种处理方式强化了强相关,弱化了弱相关或负相关,使得相关性数值更符合无标度网络特征,更具有生物意义。如果没有合适的power,一般是由于部分样品与其它样品因为某种原因差别太大导致的,可根据具体问题移除部分样品或查看后面的经验值。 - Module(模块):高度內连的基因集。在无向网络中,模块内是高度相关的基因。在有向网络中,模块内是高度正相关的基因。把基因聚类成模块后,可以对每个模块进行三个层次的分析:
1. 功能富集分析查看其功能特征是否与研究目的相符;2. 模块与性状进行关联分析,找出与关注性状相关度最高的模块;3. 模块与样本进行关联分析,找到样品特异高表达的模块。
基因富集相关文章 去东方,最好用的在线GO富集分析工具;GO、GSEA富集分析一网打进;GSEA富集分析-界面操作。其它关联后面都会提及。
- Connectivity (连接度):类似于网络中 “度” (degree)的概念。每个基因的连接度是与其相连的基因的
边属性之和。 - Module eigengene E: 给定模型的第一主成分,代表整个模型的基因表达谱。这个是个很巧妙的梳理,我们之前讲过PCA分析的降维作用,之前主要是拿来做可视化,现在用到这个地方,很好的用一个向量代替了一个矩阵,方便后期计算。(降维除了PCA,还可以看看tSNE)
- Intramodular connectivity: 给定基因与给定模型内其他基因的关联度,判断基因所属关系。
- Module membership: 给定基因表达谱与给定模型的eigengene的相关性。
- Hub gene: 关键基因 (连接度最多或连接多个模块的基因)。
- Adjacency matrix (邻接矩阵):基因和基因之间的加权相关性值构成的矩阵。
- TOM (Topological overlap matrix):把邻接矩阵转换为拓扑重叠矩阵,以降低噪音和假相关,获得的新距离矩阵,这个信息可拿来构建网络或绘制TOM图。
二、WGCNA分析流程
WGCNA根据需要计算的步骤可大致分成以下几个步骤:
1.数据前处理
2.构建加权相关性邻接矩阵
3.计算拓扑重叠矩阵
4.对基因进行层次聚类,划分模块

相关比较概念参考第二篇博客。
三、看视频(嫌麻烦倍速看)
百度云链接:
看了概念介绍之后,看代码讲解的时候,边看边运行代码。
(如果你实在是不想看,那就看后面的讲解,没有视频直接)
四、不想看视频的:
https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/
看WGCNA的教程,代码讲解一应俱全。

红色的,点过去。这个教程是基于一个小鼠疾病性状的分析。所以有些东西需要自己去改一些文件和一些参数。


所有代码都有解析的。
那么首先安装R,然后安装Rstudio。
也可以在服务器上做。
服务器名字加:8787
比如http://gs3.genek.tv:8787
前面就是服务器的名字,后面的8787就是Rstudio-server,在线服务,但是在加8787前你必须在服务器上搭建Rstudio的环境,具体步骤要看你的安装系统是什么。具体步骤CSDN上都有代码和教程,多试几次,有点看脸。
然后进入Rstudio界面是这个样子:

右下展示的PLOTS,是你绘图预览的部分。
Pakages是Rstudio的包,你已经下载了前面会有勾,下载R包可以在左上用代码,也可以在这里直接在你需要的包前点勾,但是用代码会下载相关依赖包,这里只有单个包。

(emmm提醒一下Debug这个功能,它比较抽风,有时候好用,有时候把你对的也报错,毕竟它只是一个机器不能全信,用我被别人吐槽的一句:你是人,它是机器,你决定它,不是它决定你)
现在基本了解Rstudio后,要先准备我们进行WGCNA的文件。

两个文件,一个基因表达的数据,一个是性状数据。

这个是第一个文件,注意列和行是什么,名字也要注意,特别是圈起来这里,要和性状数据一致,不然会在性状和模块合并时空。


好了,现在我们文件有了,然后开始:
第一步,安装WGCNA包:
连接到bioconductor这个网站,下载WGCNA包。
source("https://bioconductor.org/biocLite.R")
biocLite(c("AnnotationDbi", "impute","GO.db", "preprocessCore"))
site="https://mirrors.tuna.tsinghua.edu.cn/CRAN"
install.packages(c("WGCNA", "stringr", "reshape2"), repos=site)
这几句,但是它还是会有几个依赖包安不上,你看出来的结果,他会显示哪些包没有安装上,那就去bioconductor找代码,手动安装,最多只有三个包需要手动安装。



那么现在安好WGCNA后,检查安装好了没有:
library(WGCNA)看有没报错。
有就……,那就重新安?我还没遇到过。。。。。
没有接下来:


绿色框部分,关于要不要剪切看你自己一开始文件的GENEID情况。





如果聚类结果比较好,就不要这部分代码,不用切。


好的现在我们已经做好了第一步数据导入和一些预处理,下面开始构建共表达网络,有三种方法,这里只对一步法和分步法代码进行展示,大数据批量处理,感觉不太有机会用上?
首先一步法






接下来分步法:






因为可以明显发现我们的模块其实划分的很细,所以我们还要把相似的模块进行合并。那么在合并的时候我们是根据MEs值来确定的。所以我们先计算MEs值。




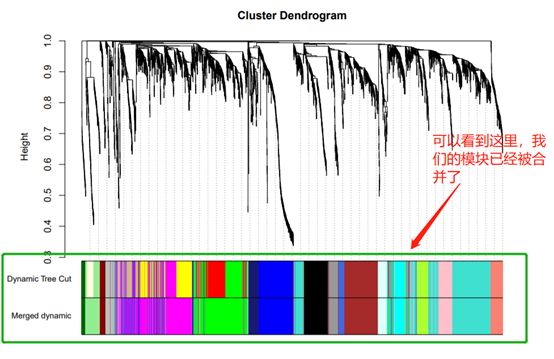
现在我们已经建立好了共表达网络,同时还将基因聚类划分了模块。
下面我们就将性状数据联合起来,确定重要的基因。



越绿代表负相关性越高,越红代表正相关性越高。
计算MM值和GS值。
GS:每个基因对应的性状的相关性,GS值越大说明这个基因和这个性状相关性越大。
MM:模块特征基因和模块关系,MM值越大说明这个特征基因对这个模块贡献越大。


接下来我们绘制GS和MM值的散点图。


网络分析结果的汇总输出
我们已经找到了与我们感兴趣的特质高度关联的模块,并通过模块成员资格度量值。现在,我们将此统计信息与基因注释合并,并编写出一个总结最重要的结果的文件,并可在标准电子表格软件(如 MS)中进行检查Excel 或打开办公室计算。


接下来我们可视化基因网络。


可视化基因网络






在这里关于后续富集分析的和如果确定hub gene 的见下一篇?
相关参考资料:
https://mp.weixin.qq.com/s/PMb2xwADvnMwaipyFXdtzQ
https://blog.csdn.net/weixin_44377608/article/details/90489878
http://www.biotrainee.com/jmzeng/html_report/d/e/e/p/i/n/WGCNA_Traits_result/index.html
http://www.bio-info-trainee.com/2297.html