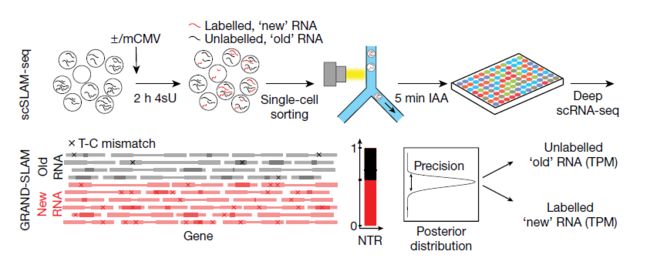
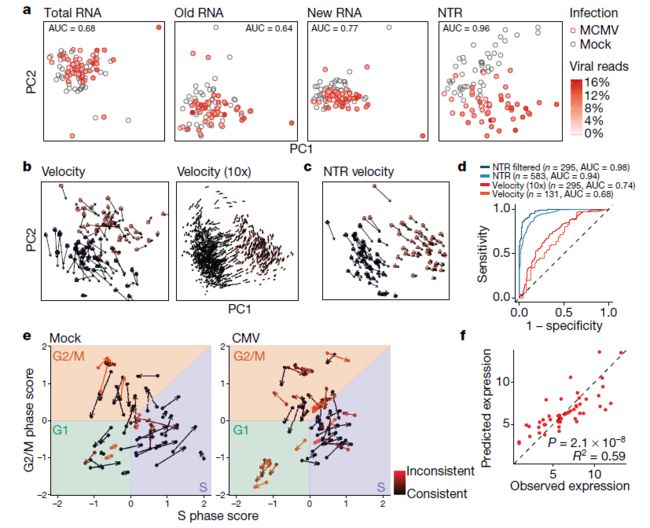
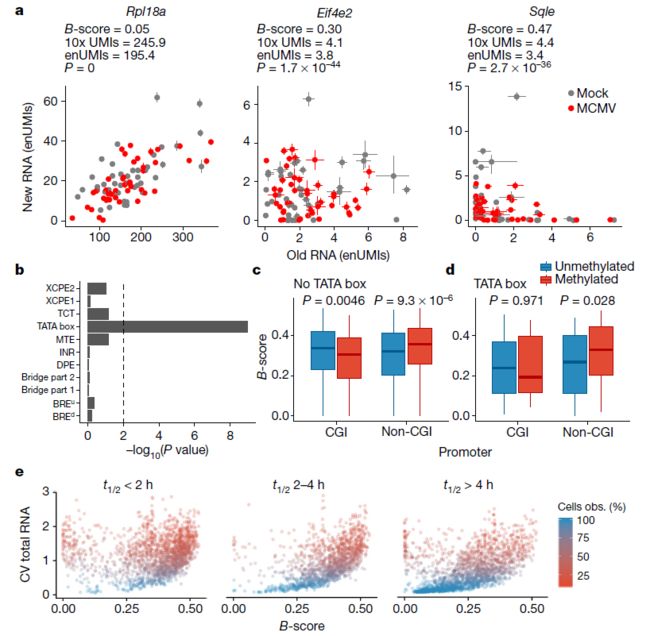
- 构建医学文献智能助手:基于 LangChain 的专业领域 RAG 系统实践
前言在当今医疗科技快速发展的时代,每天都有数以千计的医学研究成果在全球范围内发表。从临床试验报告到基础研究论文,从流行病学调查到药物研发数据,这些专业文献承载着推动医学进步的重要知识。然而,面对如此海量且专业性极强的文献资料,医疗从业者往往感到力不从心。如何在有限的时间内,准确把握文献核心价值,并将其转化为临床实践的指导?这个问题一直困扰着整个医疗行业。1.项目背景与业务价值1.1医学文献阅读的困
- python做生物信息学分析_Python从零开始第五章生物信息学①提取差异基因
吴敬欣
python做生物信息学分析
目前来说,做生物信息学的人越来越多,但是我觉得目前而言做生信的主要有三类人:老本行是做实验的,做生信可能是为了辅助研究或者是为了发paper(有非常多的临床生选择趟生信这波水)主要是做生信的,主要涵盖高通量测序数据分析,组学数据分析等等,专门从事生物学数据分析的这群人,其大部分也是本科生物狗作为强大的生力军,以调包写R,python为主。那么这群人就要熟悉看各种包的tutorial以及如何进行常规
- 用Python实现生信分析——功能预测详解
写代码的M教授
生信分析python开发语言
功能预测是生物信息学中的一项重要任务,通过分析基因或蛋白质序列的特征,推测它们的生物学功能。功能预测通常涉及多种方法,包括序列比对、基序识别、机器学习模型等。这些方法可以帮助科学家推断未知基因的功能,从而加速生物学研究的进展。1.功能预测的主要方法(1)同源性比对:通过将未知基因或蛋白质序列与数据库中的已知序列进行比对,识别出同源序列,并推测它们的功能。常用工具包括BLAST、HMMER等。(2)
- 用Python实现生信分析——序列搜索和比对工具详解
写代码的M教授
生信分析python
1.什么是序列搜索和比对工具?序列搜索和比对工具在生物信息学中用于在大型序列数据库中搜索与查询序列相似的序列,并进行比对分析。这些工具可以帮助研究人员识别与目标序列相关的已知序列,从而推测其功能、结构和进化关系。常见的序列搜索和比对工具包括:BLAST(BasicLocalAlignmentSearchTool):最常用的序列搜索工具,能够快速找到与查询序列相似的序列。FASTA:另一个常用的序列
- 如何构建知识库
追逐此刻
其他其他
构建个人知识库是一个系统化的过程,需要结合工具选择、信息管理和持续优化。以下是分步骤的实用指南,包含现代工具和方法的建议:一、明确知识库定位(Why)核心目标学习型:支持学术研究/职业发展(如医学生构建临床知识体系)创作型:支撑内容产出(如自媒体作者的选题库)项目型:管理特定领域知识(如程序员的技术栈文档)领域聚焦建议采用「T型策略」:1个深度领域+3个辅助领域(如主攻机器学习,辅修心理学/设计/
- 2025高考北京及周边理工科大学信息
东北豆子哥
高考
文章目录清华大学**1.工程与技术类(全球顶尖)****2.自然科学类****3.生命科学与医学****4.社会科学与管理类****5.人文与艺术类****6.交叉学科与新兴领域****国内学科评估参考(教育部第四轮学科评估)****选择建议**北京航空航天大学**1.航空航天类(全国顶尖)****2.信息与工程类(国内领先)****3.基础科学与工程技术****4.新兴交叉学科****5.理科与
- 大模型在坏疽及穿孔性阑尾炎预测与治疗方案制定中的应用研究
LCG元
围术期危险因子预测模型研究人工智能机器学习深度学习
目录一、引言1.1研究背景与意义1.2研究目的与创新点二、大模型技术概述2.1大模型原理与架构2.2医学领域相关应用案例三、坏疽及穿孔性阑尾炎的术前预测3.1危险因素分析3.2大模型预测模型构建3.3预测结果与临床评估四、基于预测的手术方案制定4.1手术方式选择依据4.2手术步骤与关键要点4.3案例分析五、麻醉方案确定5.1麻醉方式选择5.2麻醉药物使用5.3麻醉过程监测与管理六、术中情况监测与处
- 入选 ICML 2025!哈佛医学院等推出全球首个 HIE 领域临床思维图谱模型,神经认知结果预测任务上性能提升 15%
hyperai
在人工智能技术突飞猛进的当下,大型视觉-语言模型(LVLMs)正以惊人的速度重塑多个领域的认知边界。在自然图像与视频分析领域,这类模型依托先进的神经网络架构、海量标注数据集与强大算力支持,已能精准完成物体识别、场景解析等高阶任务。而在自然语言处理领域,LVLMs通过对TB级文本语料的学习,在机器翻译、文本摘要、情感分析等任务上达到专业级水准,其生成的学术摘要甚至能精准提炼医学文献的核心结论。然而当
- 信创时代技术栈选择与前景分析:国产替代背景下的战略路径与实践指南
猿享天开
信创开发系统安全科技创业创新开发语言
博主简介:CSDN博客专家、CSDN平台优质创作者,高级开发工程师,数学专业,10年以上C/C++,C#,Java等多种编程语言开发经验,拥有高级工程师证书;擅长C/C++、C#等开发语言,熟悉Java常用开发技术,能熟练应用常用数据库SQLserver,Oracle,mysql,postgresql等进行开发应用,熟悉DICOM医学影像及DICOM协议,业余时间自学JavaScript,Vue,
- [arXiv 2024] Medical SAM 2: Segment Medical Images as Video via Segment Anything Model 2
alfred_torres
医学图像分割SAM2
arXiv2024|MedicalSAM2:通用2D/3D医学分割新范式,“把医学图像当视频分割”论文信息标题:MedicalSAM2:SegmentMedicalImagesasVideoviaSegmentAnythingModel2作者:JiayuanZhu,AbdullahHamdi,YunliQi,YuemingJin,JundeWu单位:牛津大学、新加坡国立大学项目主页:https:/
- [CVPR 2025] 高效无监督Prompt与偏好对齐驱动的半监督医学分割
alfred_torres
prompt医学图像分割
CVPR2025|优化SAM:高效无监督Prompt与偏好对齐驱动的半监督医学分割论文信息标题:EnhancingSAMwithEfficientPromptingandPreferenceOptimizationforSemi-supervisedMedicalImageSegmentation作者:AishikKonwer,ZhijianYang,ErhanBas,CaoXiao,Pratee
- VC++实现的快速傅里叶变换频谱分析软件
直推小新
本文还有配套的精品资源,点击获取简介:基于VC++和MFC的频谱分析程序通过快速傅里叶变换(FFT)技术,将时域信号转换至频域,实现对导入文本或Excel数据的离散谱分析。用户可通过图形界面轻松导入数据,选择分析选项并查看结果。程序利用FFT高效地计算频域数据,并通过图表展示信号频率成分。此分析工具适用于音频处理、通信、医学成像和机械故障诊断等领域。1.VC++和MFC框架介绍1.1VC++的发展
- 医疗大模型深度剖析:腾讯医疗大模型案例,引领智能医疗新时代!
腾讯医疗大模型是混元大模型的医疗版。在DeepSeek爆火之前,腾讯健康已经依据医疗细分场景的具体需求,以腾讯自研的混元大模型,打造出医疗行业大模型。DeepSeek-R1发布后,腾讯健康第一时间完成了混元大模型与DeepSeek的融合。腾讯医疗大模型深度融合医学知识库与自然语言处理技术,旨在为医疗行业提供智能化的辅助解决方案。通过海量医学文献、临床指南、电子病历等专业数据训练,具备强大的医学知识
- SimpleITK——创建nrrd体素模型
bianguanyue
C#c#健康医疗算法
在介绍如何生成nrrd前,了解一下为什么医学影像上一般使用nrrd的体素模型?为什么医学影像上一般使用nrrd的体素模型?在医学影像领域,NRRD(NearlyRawRasterData)格式被广泛用于存储体素模型(如CT、MRI数据),主要基于以下技术优势:1.灵活的数据存储方式支持原始数据无损存储NRRD可直接存储未经压缩的体素数据(如16位整型、32位浮点),避免DICOM等格式
- 一[3.0]、 yolov8 工作原理
他人是一面镜子,保持谦虚的态度
车道检测研究YOLO
目录YOLOv8简介什么是YOLOv8?yaml配置文件解析YOLOv8架构图Yolov8有什么新功能?YOLO模型彻底改变了计算机视觉领域。识别物体是计算机视觉中的一项关键任务,可应用于机器人、医学成像、监控系统和自动驾驶汽车等多个领域。YOLO模型的最新版本YOLOv8是一种先进的实时物体检测框架,引起了研究界的关注。在所有流行的物体识别机器学习模型(如FasterR-CNN、SSD和Reti
- 产教融合3.0时代:数字影像产业园‘人才+产业+创新’生态闭环构建
cdsmjt
企业微信
产教融合3.0时代,数字影像产业园构建“人才+产业+创新”生态闭环,需要从以下几个关键环节入手:一、人才培养:以产业需求为导向校企深度合作:打破传统“单向输送”模式,构建双向互动机制。高校与企业共同制定人才培养方案,课程设置紧贴产业前沿技术和岗位需求。实战化教学:建设实训基地,引入真实项目,让学生在实践中掌握技能。例如,可以参照“雅职·海信人工智能医学影像三维重建生产性实训基地”模式,打造高仿真的
- 优格杂志优格杂志社优格编辑部2025年第11期部分目录
QQ296078736
人工智能
优格杂志社优格编辑部2025年第11期部分目录城市养生社区养老模式下老年人心理护理需求乌云高娃1-3走进超声医学的奇妙世界:揭秘超声技术的多样性胡丽丽4-6做有温度的产科护理,筑牢母婴安全防线鲁娜李襄君7精准翻身干预:降低压疮发生率的新方法陈思8月经紊乱与潜在疾病的关联马占兰9师者说让体育课成为生命成长的摇篮杜俊义10读说写教学模式在英语课堂如何人文化实施李刚强11巧借小学数学教学,培育学生数学思
- 武魂杂志武魂杂志社武魂编辑部2025年第1期部分目录
QQ296078736
人工智能
武魂杂志社编辑部2025年第1期部分目录武术大视野体育核心素养视域下武术教学的价值意蕴与实践路径张向楠1-3AI技术在青少年跆拳道教学中的应用刘晓珊王子伦4-6福州市拳击俱乐部教练队伍发展现状与对策分析李怡婕王丹7-9"双减"背景下小学跆拳道课后延时服务发展的实践策略林文烨10-12课程思政元素融入高校医学生太极拳课程的教学策略探究陈明晓13-15人工智能在高校八段锦教学中的应用与实施策略张亚贵采
- 博弈论概述
C7211BA
博弈论
博弈论(GameTheory)是研究理性决策者在策略互动中如何行动和决策的数学理论。它广泛应用于经济学、政治学、生物学、计算机科学等领域。以下是博弈论的主要思想和核心概念:1.核心思想博弈论的核心是分析多个参与者(玩家)在相互依赖的情境中如何做出最优决策,即每个人的收益不仅取决于自己的选择,还取决于他人的选择。主要特点包括:策略互动:玩家的决策相互影响。理性假设:玩家追求自身利益最大化(理性人假设
- vtk和opencv和opengl直接的区别是什么?
only-lucky
opencv人工智能计算机视觉
简介VTK、OpenCV和OpenGL是三个在计算机图形学、图像处理和可视化领域广泛使用的工具库,但它们在功能、应用场景和底层技术上存在显著差异。以下是它们的核心区别和特点对比:1.核心功能与定位工具核心功能主要应用领域VTK(VisualizationToolkit)三维可视化&科学计算,提供高级渲染、体绘制、交互式可视化医学影像、地质建模、流体力学仿真OpenCV(OpenSourceComp
- 中医学习小课堂-源代码
钰清雅
游戏python开发语言
中医学习小课堂-创作思路1.设计理念与核心目标寓教于乐:将中医药知识融入游戏化学习,通过“诊断开方”的互动形式激发学习兴趣知识结构化:建立完整的中药知识体系(药性、归经、功效、分类)和病症辨证模型实践导向:模拟真实中医诊疗流程(望闻问切→辨证→开方→验证)分层学习:提供“学习模式”和“诊疗模式”满足不同基础的学习者需求2.功能架构设计graphTDA[核心系统]-->B[药材库]A-->C[患者系
- UNet改进(5):线性注意力机制(Linear Attention)-原理详解与代码实现
摸鱼许可证
人工智能计算机视觉
引言在计算机视觉领域,UNet架构因其在图像分割任务中的卓越表现而广受欢迎。近年来,注意力机制的引入进一步提升了UNet的性能。本文将深入分析一个结合了线性注意力机制的UNet实现,探讨其设计原理、代码实现以及在医学图像分割等任务中的应用潜力。UNet架构概述UNet最初由Ronneberger等人提出,主要用于生物医学图像分割。其独特的U形结构由编码器(下采样路径)和解码器(上采样路径)组成,通
- Java医学图像处理系统实战源码剖析
好学的Jack
本文还有配套的精品资源,点击获取简介:本项目详细介绍了基于Java的医学图像处理系统,通过使用Java提供的图像处理库和多线程技术,实现了医疗图像的读取、预处理、分析、分割、存储及报告生成等关键功能。系统不仅支持多种图像格式和数据库集成,还考虑了用户界面设计和数据安全性,为医疗领域的图像分析需求提供了解决方案。学生和开发者可通过源码学习和实践,深入了解如何构建一个功能全面的医学图像处理平台。1.J
- 掌握Python:从基础到AI开发的首选语言
博主简介:CSDN博客专家、CSDN平台优质创作者,高级开发工程师,数学专业,10年以上C/C++,C#,Java等多种编程语言开发经验,拥有高级工程师证书;擅长C/C++、C#等开发语言,熟悉Java常用开发技术,能熟练应用常用数据库SQLserver,Oracle,mysql,postgresql等进行开发应用,熟悉DICOM医学影像及DICOM协议,业余时间自学JavaScript,Vue,
- 浙江大学公开课第三季:AI+BME,迈向智慧医疗健康——浙大的探索与实践(附PDF下载)
大模型应用
人工智能自然语言处理LLM知识图谱RAG大模型程序员
浙江大学:《AI+BME,迈向智慧医疗健康——浙大的探索与实践》(完整版.pdf下载方式见文末)该报告是关于浙江大学在生物医学工程(BME)与人工智能(AI)结合领域的探索与实践,重点介绍了浙大在智慧医疗健康领域的研究成果和未来发展方向。文章由浙江大学生物医学工程与仪器科学学院的林辉教授撰写。1.AI与BME的结合AI的发展历程:文章回顾了人工智能的发展历史,从1956年达特茅斯会议标志AI诞生,
- 半导体材料仿真:量子点材料仿真_(20).量子点材料在生物医学中的应用案例
kkchenkx
信号仿真2量子计算信息可视化信号处理人工智能数据分析
量子点材料在生物医学中的应用案例在上一节中,我们讨论了量子点材料的基本性质和合成方法。本节将重点介绍量子点材料在生物医学领域的应用案例,包括生物标记、成像、药物递送和光疗等方面。通过这些案例,我们将深入了解量子点材料在生物医学中的独特优势和潜在应用。1.量子点在生物标记中的应用1.1生物标记的基本原理生物标记是指使用特定的化学或物理方法对生物分子、细胞或组织进行标记,以便于观察和分析。量子点由于其
- AI人工智能加持,联邦学习医疗数据共享方案全解析
AI学长带你学AI
CS人工智能网络ai
AI人工智能加持,联邦学习医疗数据共享方案全解析关键词:联邦学习、医疗数据共享、隐私保护、人工智能、多方安全计算摘要:医疗数据是医学研究和临床决策的“黄金资源”,但患者隐私保护与数据孤岛问题却像两道高墙,阻碍着医疗AI的发展。本文将以“联邦学习”这一AI核心技术为钥匙,带您深入理解如何在不泄露原始数据的前提下,实现跨医院、跨机构的医疗数据共享与联合建模。我们将从生活场景出发,用“厨师合作研发新菜”
- 基于深度学习的智能图像语义分割系统:技术与实践
Blossom.118
机器学习与人工智能深度学习人工智能python分类音视频机器学习sklearn
前言图像语义分割是计算机视觉领域中的一个重要任务,其目标是将图像中的每个像素分配到预定义的语义类别中。这一技术在自动驾驶、医学影像分析、机器人视觉等多个领域有着广泛的应用。近年来,深度学习技术,尤其是卷积神经网络(CNN)及其变体,为图像语义分割带来了显著的改进。本文将详细介绍基于深度学习的智能图像语义分割系统的原理、实现方法以及实际应用案例。一、图像语义分割的基本概念1.1什么是图像语义分割?图
- 在VTK中捕捉体绘制图像并实时图像处理
点PY
三维渲染图像处理人工智能VTK
0.概要这段代码实现了一个高级的医学图像可视化系统,主要特点包括双窗口交互式体绘制、图像后处理和实时同步。1.核心功能架构主窗口:3D体绘制视图(GPU加速的体积渲染)副窗口:2D截图视图(带高斯模糊后处理)交互机制:副窗口的交互操作会实时影响主窗口的3D视图2.关键组件分析2.1自定义交互器(CustomInteractorStyle)classCustomInteractorStyle:
- Patch Position Embedding (PPE) 在医疗 AI 中的应用编程分析
Allen_Lyb
数智化教程(第二期)embedding人工智能机器学习健康医疗
一、PPE的核心原理与医疗场景适配性位置编码的本质需求在医疗影像(如CT、MRI、病理切片)中,Transformer需要将图像划分为若干Patch并作为序列输入。但如果不注入空间信息,模型难以区分同一病灶在不同坐标的语义差异。传统的绝对位置编码(如SinusoidalPE)对等距网格有效,却无法灵活适配病灶大小多变、图像分辨率不一的医学场景。PatchPositionEmbedding(PPE)
- java短路运算符和逻辑运算符的区别
3213213333332132
java基础
/*
* 逻辑运算符——不论是什么条件都要执行左右两边代码
* 短路运算符——我认为在底层就是利用物理电路的“并联”和“串联”实现的
* 原理很简单,并联电路代表短路或(||),串联电路代表短路与(&&)。
*
* 并联电路两个开关只要有一个开关闭合,电路就会通。
* 类似于短路或(||),只要有其中一个为true(开关闭合)是
- Java异常那些不得不说的事
白糖_
javaexception
一、在finally块中做数据回收操作
比如数据库连接都是很宝贵的,所以最好在finally中关闭连接。
JDBCAgent jdbc = new JDBCAgent();
try{
jdbc.excute("select * from ctp_log");
}catch(SQLException e){
...
}finally{
jdbc.close();
- utf-8与utf-8(无BOM)的区别
dcj3sjt126com
PHP
BOM——Byte Order Mark,就是字节序标记 在UCS 编码中有一个叫做"ZERO WIDTH NO-BREAK SPACE"的字符,它的编码是FEFF。而FFFE在UCS中是不存在的字符,所以不应该出现在实际传输中。UCS规范建议我们在传输字节流前,先传输 字符"ZERO WIDTH NO-BREAK SPACE"。这样如
- JAVA Annotation之定义篇
周凡杨
java注解annotation入门注释
Annotation: 译为注释或注解
An annotation, in the Java computer programming language, is a form of syntactic metadata that can be added to Java source code. Classes, methods, variables, pa
- tomcat的多域名、虚拟主机配置
g21121
tomcat
众所周知apache可以配置多域名和虚拟主机,而且配置起来比较简单,但是项目用到的是tomcat,配来配去总是不成功。查了些资料才总算可以,下面就跟大家分享下经验。
很多朋友搜索的内容基本是告诉我们这么配置:
在Engine标签下增面积Host标签,如下:
<Host name="www.site1.com" appBase="webapps"
- Linux SSH 错误解析(Capistrano 的cap 访问错误 Permission )
510888780
linuxcapistrano
1.ssh -v
[email protected] 出现
Permission denied (publickey,gssapi-keyex,gssapi-with-mic,password).
错误
运行状况如下:
OpenSSH_5.3p1, OpenSSL 1.0.1e-fips 11 Feb 2013
debug1: Reading configuratio
- log4j的用法
Harry642
javalog4j
一、前言: log4j 是一个开放源码项目,是广泛使用的以Java编写的日志记录包。由于log4j出色的表现, 当时在log4j完成时,log4j开发组织曾建议sun在jdk1.4中用log4j取代jdk1.4 的日志工具类,但当时jdk1.4已接近完成,所以sun拒绝使用log4j,当在java开发中
- mysql、sqlserver、oracle分页,java分页统一接口实现
aijuans
oraclejave
定义:pageStart 起始页,pageEnd 终止页,pageSize页面容量
oracle分页:
select * from ( select mytable.*,rownum num from (实际传的SQL) where rownum<=pageEnd) where num>=pageStart
sqlServer分页:
- Hessian 简单例子
antlove
javaWebservicehessian
hello.hessian.MyCar.java
package hessian.pojo;
import java.io.Serializable;
public class MyCar implements Serializable {
private static final long serialVersionUID = 473690540190845543
- 数据库对象的同义词和序列
百合不是茶
sql序列同义词ORACLE权限
回顾简单的数据库权限等命令;
解锁用户和锁定用户
alter user scott account lock/unlock;
//system下查看系统中的用户
select * dba_users;
//创建用户名和密码
create user wj identified by wj;
identified by
//授予连接权和建表权
grant connect to
- 使用Powermock和mockito测试静态方法
bijian1013
持续集成单元测试mockitoPowermock
实例:
package com.bijian.study;
import static org.junit.Assert.assertEquals;
import java.io.IOException;
import org.junit.Before;
import org.junit.Test;
import or
- 精通Oracle10编程SQL(6)访问ORACLE
bijian1013
oracle数据库plsql
/*
*访问ORACLE
*/
--检索单行数据
--使用标量变量接收数据
DECLARE
v_ename emp.ename%TYPE;
v_sal emp.sal%TYPE;
BEGIN
select ename,sal into v_ename,v_sal
from emp where empno=&no;
dbms_output.pu
- 【Nginx四】Nginx作为HTTP负载均衡服务器
bit1129
nginx
Nginx的另一个常用的功能是作为负载均衡服务器。一个典型的web应用系统,通过负载均衡服务器,可以使得应用有多台后端服务器来响应客户端的请求。一个应用配置多台后端服务器,可以带来很多好处:
负载均衡的好处
增加可用资源
增加吞吐量
加快响应速度,降低延时
出错的重试验机制
Nginx主要支持三种均衡算法:
round-robin
l
- jquery-validation备忘
白糖_
jquerycssF#Firebug
留点学习jquery validation总结的代码:
function checkForm(){
validator = $("#commentForm").validate({// #formId为需要进行验证的表单ID
errorElement :"span",// 使用"div"标签标记错误, 默认:&
- solr限制admin界面访问(端口限制和http授权限制)
ronin47
限定Ip访问
solr的管理界面可以帮助我们做很多事情,但是把solr程序放到公网之后就要限制对admin的访问了。
可以通过tomcat的http基本授权来做限制,也可以通过iptables防火墙来限制。
我们先看如何通过tomcat配置http授权限制。
第一步: 在tomcat的conf/tomcat-users.xml文件中添加管理用户,比如:
<userusername="ad
- 多线程-用JAVA写一个多线程程序,写四个线程,其中二个对一个变量加1,另外二个对一个变量减1
bylijinnan
java多线程
public class IncDecThread {
private int j=10;
/*
* 题目:用JAVA写一个多线程程序,写四个线程,其中二个对一个变量加1,另外二个对一个变量减1
* 两个问题:
* 1、线程同步--synchronized
* 2、线程之间如何共享同一个j变量--内部类
*/
public static
- 买房历程
cfyme
2015-06-21: 万科未来城,看房子
2015-06-26: 办理贷款手续,贷款73万,贷款利率5.65=5.3675
2015-06-27: 房子首付,签完合同
2015-06-28,央行宣布降息 0.25,就2天的时间差啊,没赶上。
首付,老婆找他的小姐妹接了5万,另外几个朋友借了1-
- [军事与科技]制造大型太空战舰的前奏
comsci
制造
天气热了........空调和电扇要准备好..........
最近,世界形势日趋复杂化,战争的阴影开始覆盖全世界..........
所以,我们不得不关
- dateformat
dai_lm
DateFormat
"Symbol Meaning Presentation Ex."
"------ ------- ------------ ----"
"G era designator (Text) AD"
"y year
- Hadoop如何实现关联计算
datamachine
mapreducehadoop关联计算
选择Hadoop,低成本和高扩展性是主要原因,但但它的开发效率实在无法让人满意。
以关联计算为例。
假设:HDFS上有2个文件,分别是客户信息和订单信息,customerID是它们之间的关联字段。如何进行关联计算,以便将客户名称添加到订单列表中?
&nbs
- 用户模型中修改用户信息时,密码是如何处理的
dcj3sjt126com
yii
当我添加或修改用户记录的时候对于处理确认密码我遇到了一些麻烦,所有我想分享一下我是怎么处理的。
场景是使用的基本的那些(系统自带),你需要有一个数据表(user)并且表中有一个密码字段(password),它使用 sha1、md5或其他加密方式加密用户密码。
面是它的工作流程: 当创建用户的时候密码需要加密并且保存,但当修改用户记录时如果使用同样的场景我们最终就会把用户加密过的密码再次加密,这
- 中文 iOS/Mac 开发博客列表
dcj3sjt126com
Blog
本博客列表会不断更新维护,如果有推荐的博客,请到此处提交博客信息。
本博客列表涉及的文章内容支持 定制化Google搜索,特别感谢 JeOam 提供并帮助更新。
本博客列表也提供同步更新的OPML文件(下载OPML文件),可供导入到例如feedly等第三方定阅工具中,特别感谢 lcepy 提供自动转换脚本。这里有导入教程。
- js去除空格,去除左右两端的空格
蕃薯耀
去除左右两端的空格js去掉所有空格js去除空格
js去除空格,去除左右两端的空格
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>&g
- SpringMVC4零配置--web.xml
hanqunfeng
springmvc4
servlet3.0+规范后,允许servlet,filter,listener不必声明在web.xml中,而是以硬编码的方式存在,实现容器的零配置。
ServletContainerInitializer:启动容器时负责加载相关配置
package javax.servlet;
import java.util.Set;
public interface ServletContainer
- 《开源框架那些事儿21》:巧借力与借巧力
j2eetop
框架UI
同样做前端UI,为什么有人花了一点力气,就可以做好?而有的人费尽全力,仍然错误百出?我们可以先看看几个故事。
故事1:巧借力,乌鸦也可以吃核桃
有一个盛产核桃的村子,每年秋末冬初,成群的乌鸦总会来到这里,到果园里捡拾那些被果农们遗落的核桃。
核桃仁虽然美味,但是外壳那么坚硬,乌鸦怎么才能吃到呢?原来乌鸦先把核桃叼起,然后飞到高高的树枝上,再将核桃摔下去,核桃落到坚硬的地面上,被撞破了,于是,
- JQuery EasyUI 验证扩展
可怜的猫
jqueryeasyui验证
最近项目中用到了前端框架-- EasyUI,在做校验的时候会涉及到很多需要自定义的内容,现把常用的验证方式总结出来,留待后用。
以下内容只需要在公用js中添加即可。
使用类似于如下:
<input class="easyui-textbox" name="mobile" id="mobile&
- 架构师之httpurlconnection----------读取和发送(流读取效率通用类)
nannan408
1.前言.
如题.
2.代码.
/*
* Copyright (c) 2015, S.F. Express Inc. All rights reserved.
*/
package com.test.test.test.send;
import java.io.IOException;
import java.io.InputStream
- Jquery性能优化
r361251
JavaScriptjquery
一、注意定义jQuery变量的时候添加var关键字
这个不仅仅是jQuery,所有javascript开发过程中,都需要注意,请一定不要定义成如下:
$loading = $('#loading'); //这个是全局定义,不知道哪里位置倒霉引用了相同的变量名,就会郁闷至死的
二、请使用一个var来定义变量
如果你使用多个变量的话,请如下方式定义:
. 代码如下:
var page
- 在eclipse项目中使用maven管理依赖
tjj006
eclipsemaven
概览:
如何导入maven项目至eclipse中
建立自有Maven Java类库服务器
建立符合maven代码库标准的自定义类库
Maven在管理Java类库方面有巨大的优势,像白衣所说就是非常“环保”。
我们平时用IDE开发都是把所需要的类库一股脑的全丢到项目目录下,然后全部添加到ide的构建路径中,如果用了SVN/CVS,这样会很容易就 把
- 中国天气网省市级联页面
x125858805
级联
1、页面及级联js
<%@ page language="java" import="java.util.*" pageEncoding="UTF-8"%>
<!DOCTYPE HTML PUBLIC "-//W3C//DTD HTML 4.01 Transitional//EN">
&l