一般来讲,对于较为复杂的基因组,我们通常会在基因组正式组装之前进行k-mer分析,以评估基因组杂合度、重复序列比例,并初步预估基因组大小,以及测序深度等,为后续基因组的正式组装提供参考信息。k-mer分析简介可参见:http://blog.sciencenet.cn/blog-3406804-1162384.html
此处给大家介绍一款可以进行k-mer分析及基因组大小评估的工具,KmerGenie。KmerGenie的最大优点在于可以实现在多个预设k-mer下的自动分析,除了进行常规的k-mer频数统计之外,还能够基于不同k-mer自动计算基因组大小,并为基因组组装评估一个最佳组装k-mer数值作为备选。
本文中的测试数据以及结果示例的百度盘链接如下,均无提取码。
测试数据:https://pan.baidu.com/s/1NFvRDs3kZsh3VK7ukv1svA
示例结果:https://pan.baidu.com/s/1lRmn_8qFGBbbwvbQPjmZbg
KmerGenie下载安装
进入KmerGenie网站(http://kmergenie.bx.psu.edu/),点击“kmergenie-1.7051.tar.gz”即可下载压缩包。
此外对于其它可查看项:点击“sample report”后可查看一份简版示例报告,方便我们在正式运行KmerGenie之前提前了解结果样式;“README”为安装及使用说明。

KmerGenie的安装需要python(>=2.7),分析功能的实现除python外还需要依赖R等环境的支持。

以上为“README”的部分截图,所需的环境配置好后,在shell命令中解压安装包并安装。
#安装
tar -xzvf kmergenie-1.7051.tar.gz && cd kmergenie-1.7051
python setup.py install
#可为所有文件(包括主程序 kmergenie)添加可执行权限
chmod -R 755 *
#更改指令方便今后的书写,可直接在 ~/.bashrc 中修改
#例如 kmergenie 的绝对路径为“/home/my/software/kmergenie-1.7051/ kmergenie”
alias kmergenie='/home/my/software/kmergenie-1.7051/kmergenie'
#修改后只需输入“kmergenie”即可实现“/home/my/software/kmergenie-1.7051/ kmergenie”的功能
安装完毕后,我们可首先使用“kmergenie -h”查看KmerGenie的运行参数,并检测KmerGenie是否安装成功。
kmergenie -h
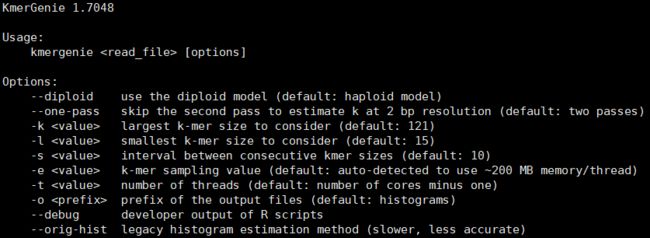
一般使用时关注几个重要参数即可。--diploid为二倍体模式,默认不使用,即单倍体模式;-k,设定最大k-mer值,默认121,最大阈值121;-l,设定最小k-mer值,默认15,最小阈值15;-s,设定k-mer取值间隔,默认在最小值~最大值之间以10为一取值间隔;-t,程序运行线程数;-e,程序运行分配内存。
KmerGenie运行示例及结果说明
单倍体模式测试
这里使用百度盘链接中测试数据演示软件使用过程。
首先我们将fastq格式的测序数据的绝对路径写入至一个文档中(见网盘附件“fastq_list.txt”,假设fastq文件位于“/home/my/data”)。输入fastq文件为经过碱基校正及reads去重后的clean data,即网盘中的“Bacillus_subtilis.filt_R1.fastq.gz / Bacillus_subtilis.filt_R2.fastq.gz”,fastq格式或gzip压缩后的fastq.gz格式均可识别。

之后运行运行KmerGenie,示例命令如下。
#示例中,“fastq_list.txt”在当前路径下
mkdir result_haploid
kmergenie fastq_list.txt -o ./result_haploid/haploid -l 15 -k 105 -s 10 -t 4 > haploid.log1.txt 2> haploid.log2.txt
默认单倍体模式,以k-mer长度15为起始,121为终止,10为间隔逐一测试;程序运行线程数4。结果输出在当前路径下的“result_haploid”中。“haploid.log1.txt”和“haploid.log2.txt”分别为程序运行时的正确/错误输出日志。
运行完毕后即可查看“result_haploid”中的结果。
我们可首先关注结果报告“report.html”。对于上述运行示例,其结果报告的内容简要如下(可见网盘附件文件夹“result_haploid”)。
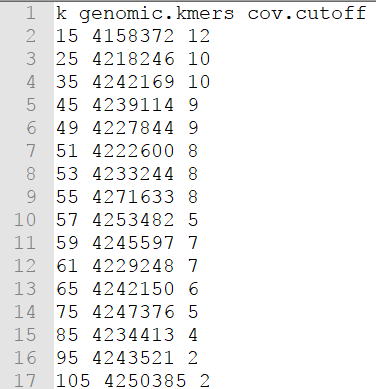
报告开头以折线图的形式展示出在每种长度k-mer下,估算的基因组大小,同时给出了“最佳k-mer”选择数值(其实就是将评估基因组总大小最高的那个k-mer值判定为“最佳k-mer”,为基因组组装时k-mer的选择提供参考)。
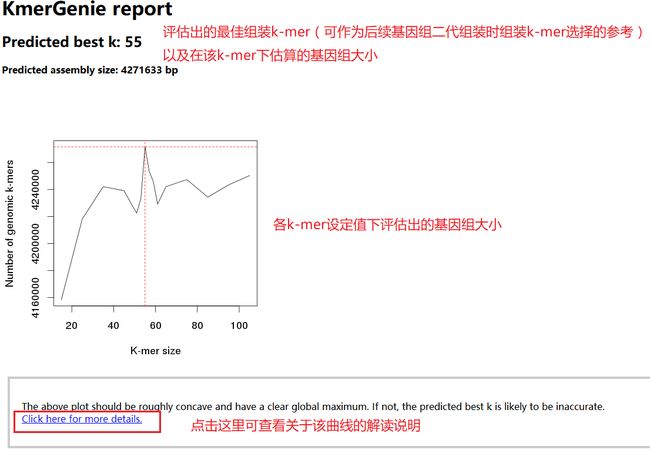
报告中部给出了曲线的详细说明,包括最佳k-mer的评估规则,以及当测序深度足够高时的k-mer选择等。
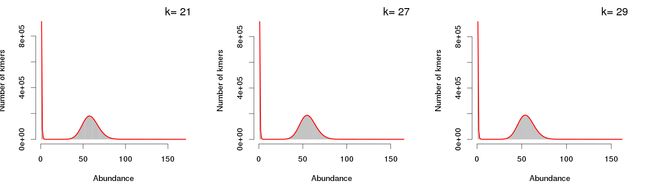
报告的下方给出了每种k-mer的频数分布图,在基因组的k-mer中可根据该图判定基因组杂合度或重复序列比例。

然后再查看“result_haploid”中的其它结果文件,其它文件均为报告结果“report.html”的配置文件,如无特殊需要(例如将其中的某个k-mer频数分布列表拿出,使用其它软件进行额外的分析),无需单独查看。
查看“*.histo”中的内容(下图左),为统计得到的k-mer的频数分布列表,即为上述展示的各k-mer设定值下的k-mer曲线的作图数据。其中,第一列为k-mer深度,即各k-mer的出现频数;第二列为出现该频数的k-mer片段总数。根据k-mer频数分布信息我们可估算基因组大小、重复序列比例、测序覆盖度等,此处需要借助其它软件读入k-mer频数列表文件进行统计计算,因此不再多说。
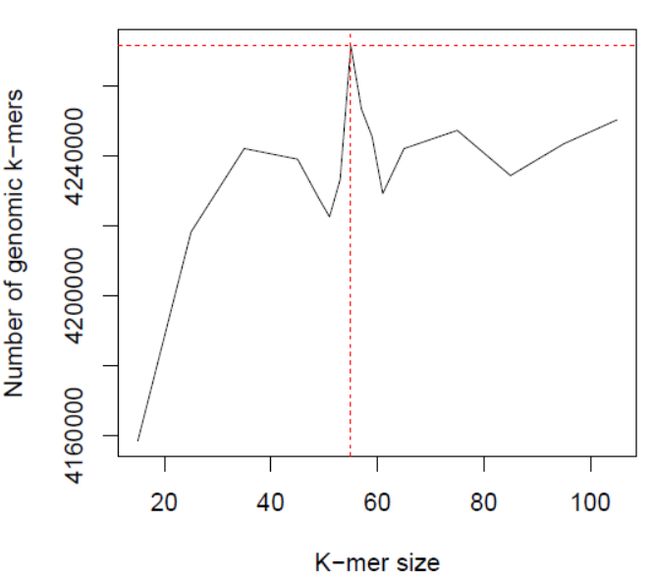
“*.histo.pdf”即为k-mer频数分布曲线(下图右),使用“*.histo”中的数据绘制,两个文件的名称一一对应。次外,我们也可基于“*.histo”中的数据自行绘制k-mer曲线展示,不再多说了。

同样地,“*.dat”与“.dat.pdf”相对应,记录了在各k-mer设定值下进行k-mer分析后所评估出的基因组大小。
二倍体模式测试
若测序物种的基因组高杂合(如通过k-mer曲线判断,例如曲线中存在杂合峰),我们可考虑运行二倍体模式,此时相较于单倍体模式的运行结果会更佳。当然,若测序物种的基因组仅为简单基因组(无杂合),则是不能使用二倍体模式运行的,强行运行二倍体模式不会得到有用的结果的。
由于网盘中所提供测序数据的来源物种,其基因组并非杂合基因组,因此不再使用该数据运行二倍体模式。本人也比较懒,不想再找杂合基因组的测序数据了......因此以下仅为演示软件使用,提供的示例运行命令。
与上述过程一致,假设此处存在测序数据“test_R1.fastq / test_R2.fastq”,并且已知该测序物种的基因组存在杂合区,首先我们将测试数据的绝对路径写入至一个文档中(假设fastq或fastq.gz文件位于“/home/my/data”)。
之后运行运行KmerGenie,示例命令如下。
#示例中,“fastq_list.txt”在当前路径下
mkdir result_diploid
kmergenie fastq_list.txt -o ./result_diploid/diploid -l 15 -k 105 -s 10 -t 4 --diploid >diploid.log1.txt 2>diploid.log2.txt
--diploid,使用二倍体模式;视情况选择合适的参数,例如此处我们以k-mer=15为起始,121为终止,10为间隔;程序运行线程数4。结果输出在当前路径下的“result_diploid”中。“diploid.log1.txt”和“diploid.log2.txt”分别为程序运行时的正确/错误输出日志。
运行完毕后即可查看“result_diploid”中的结果。同样地,我们可主要关注结果报告“report.html”即可。
以下从其它地方找了某杂合基因组物种的KmerGenie分析结果,作为对结果的解读。
同样地,报告报告开头以折线图的形式展示出在每种长度k-mer下,估算的基因组大小。事实上,二倍体运行模式下所得的基因组大小评估曲线与单倍体模式下的截然不同。大家可以自行找个杂合基因组物种的测序数据测试下。
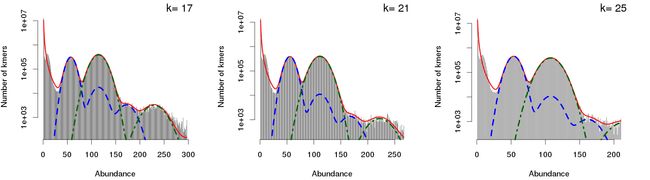
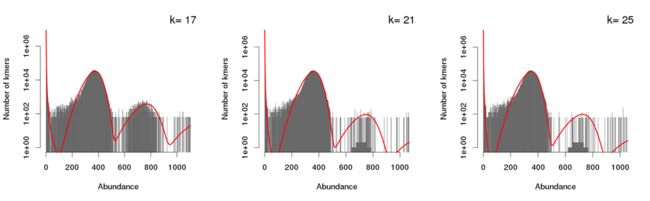
报告的下方给出了每种k-mer的频数分布图,使用了二倍体运行模式后,与单倍体运行模式下所得结果相比在展示方式上多了一些内容。例如下图(上)为二倍体运行模式下的k-mer曲线,红色曲线为观测到的k-mer曲线;蓝色曲线为杂合的k-mer曲线;绿色曲线为纯合的k-mer曲线。下图(下)为单倍体运行模式下单k-mer曲线形状,常规单曲线展示方式。
KmerGenie的其它内容
关于KmerGenie的k-mer曲线展示方式
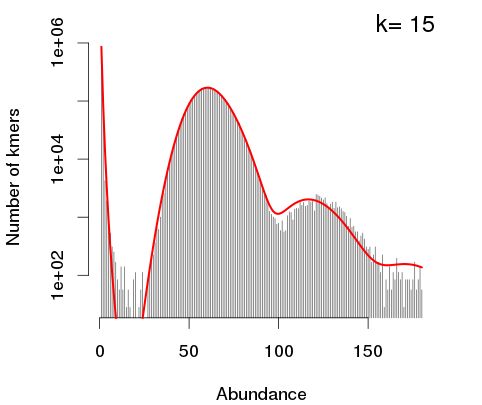
这里需要注意一点:KmerGenie软件默认将k-mer频数曲线的纵坐标进行了log10转化,因此该软件默认绘制的k-mer曲线与常规k-mer曲线(纵坐标未进行对数转化的)在视觉直观上存在差异(不注意的话,可能会引起一定的误解)。以上图为例,我们可以直观地看到纵坐标数值取了log10。
若想让KmerGenie展示原始纵坐标,即不对k-mer数目进行对数转化,可在KmerGenie的作图脚本(在KmerGenie 程序所在路径中查找,“kmergenie-1.7051/scripts/plot_histogram.r”)中进行修改。在“plot_histogram.r”中定位至第110行,将“log='y'”去掉即可。

↓↓

替换命令后,使用单倍体模式测试,KmerGenie运行参数同上述,k-mer频数曲线结果如下。
结果中曲线峰值不是很明显。主要原因由于在二代测序中因测序错误的存在会带来许多低出现频数的k-mer,而这些k-mer的总数目占据了相当一部分的比例,会对结果观测产生极大影响(在上图中,Abundance = 1、2等起始位置处出现很高的k-mer数目,使得图中曲线峰值很难分辨)。估计KmerGenie也是考虑了这一点,因此默认取了log转化,以降低测序错误率所带来的影响。若不进行log转化,为了增强结果的可视化,通常情况下我们会考虑屏蔽了低Abundance的区域。
屏蔽区域视情况而定,继续使用本示例展示的话,我们可考虑屏蔽Abundance < 5的区域。

替换命令后再次测试,这里的k-mer曲线就与我们平时见到的k-mer曲线一致了。

同样地,替换命令后,二倍体模式数据绘图展示效果也会更直观。此处不再展示了。
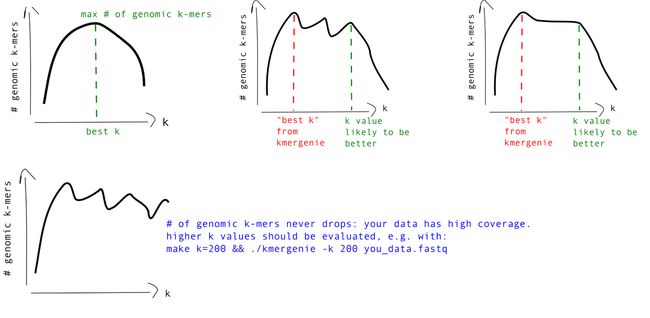
关于基因组二代组装可选k-mer取值的评估
KmerGenie评估出了一个“最佳k-mer”,为后续使用该测序数据进行基因组正式组装时所选的k-mer取值提供了参考。如下示例,从其它地方找了一个KmerGenie运行结果,而非本文中的测试数据所得结果,这里best k = 43。虽然就是将评估基因组总大小最高的那个k-mer值判定为“最佳k-mer”,若测序样本无污染的情况下,使用该k-mer值进行基因组组装的结果还是蛮可观的。
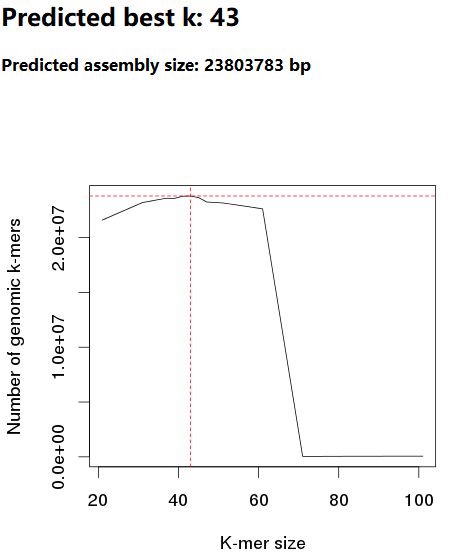
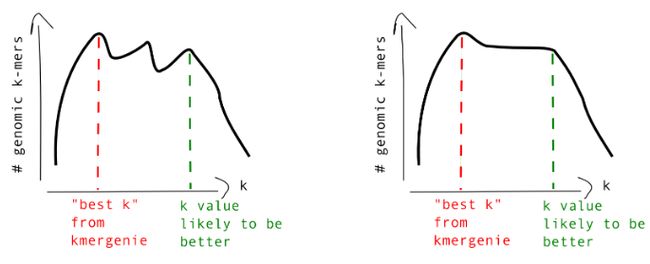
此外,根据KmerGenie所得的基因组大小评估曲线,我们还可从中选取其它较为可靠的k-mer值进行基因组组装,就如说明文档中所提及的那样:
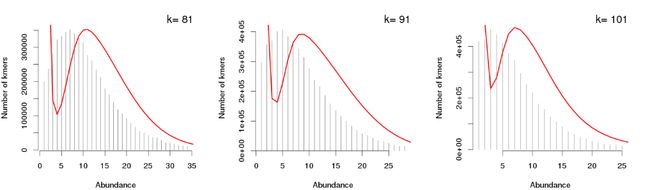
根据KmerGenie所得的k-mer频数曲线,我们还可获知一些其它的信息。比如说,在基因组组装时,k-mer的取值受测序深度的影响,若测序深度越高,我们可选择更高的k-mer进行尝试组装,以得到更长更完整的contigs序列;但若在低深度测序模式下使用较高的k-mer进行组装时,就会引入较高的错误率。
如下示例,某数据的k-mer曲线图(非log转化形式),在高k-mer设定值下,左侧由于测序错误所导致的k-mer数目并未下降至最低后即产生了上升趋势,说明使用这些k-mer将会带来高错误率,不建议使用。
本文转自刘尧科学网博客。
链接地址:http://blog.sciencenet.cn/blog-3406804-1159967.html