肿瘤抗原
癌细胞突变生成新的被免疫系统识别成非自身部分的肽链[1]。通常源于单核苷酸突变,极少部分源自多个基因变异事件。(Single mutations generate 99.8% of unique pMHCs while 0.2% result from distinct mutations in different genetic loci yielding identical peptides (Figure 5A))[2]
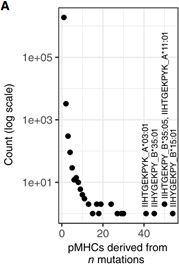
肿瘤抗原负荷(Neoantigen load/burden)
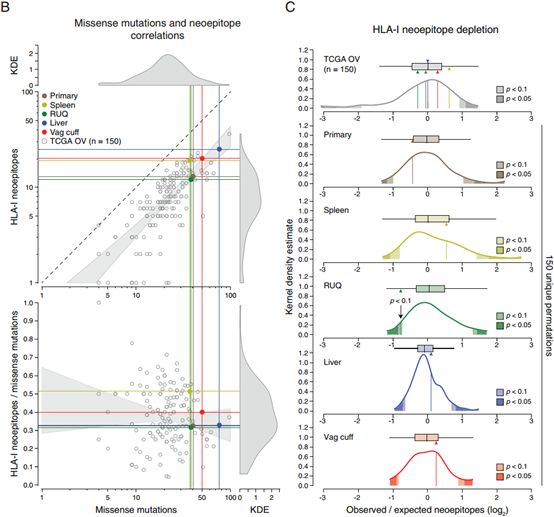
肿瘤抗原负荷通常与突变负荷、HLA类型结合来针对不同肿瘤类型或临床结果进行分析,也可以分别单独分析。可以使用CiberSort得到免疫细胞比例,再联系肿瘤抗原负荷。(The tumors with the highest mutation and neoepitope loads for both HLA class I and HLA class II were the liver and vaginal cuff, which also had the highest number of missense mutations (Figure 4B)).[3]
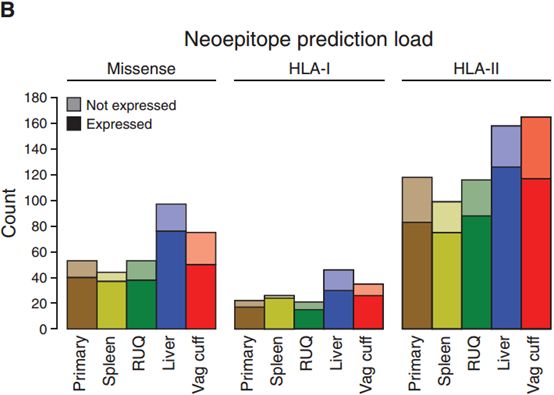
肿瘤抗原消耗(neoepitope depletion)
肿瘤抗原消耗通常作为一个佐证因素来验证肿瘤抗原消耗与疾病发展状态关系,通常的关系是“肿瘤抗原消耗与肿瘤退化或稳定相关”。 we further interrogated the neoepitope landscape by analyzing potential evidence of neoepitope depletion using an approach adopted from a report analyzing TCGA data. Relative to the other samples from the patient, the regressing RUQ tumor showed a consistent—yet non-significant—tendency of neoepitope depletion (Figure S4B and S4C). This result is in line with a recent report showing neoepitope depletion in tumors with higher levels of immune signatures in colorectal cancer (Davoli et al., 2017). [3]
共享肿瘤抗原(Shared Neoantigen)
可以结合肿瘤类型与临床结果,预后结果,观察是否与共享肿瘤抗原相关。We also investigated whether there were shared neoepitopes or mutations present in the RUQ (regressing) and liver (stable) metastases alone, i.e., not present in the other tumors. No shared mutations between RUQ and liver alone were detected (Figure S4A); therefore, it did not appear that a shared neoepitope or mutation alone explained the behavior of the non-progressing tumor sites.[3]

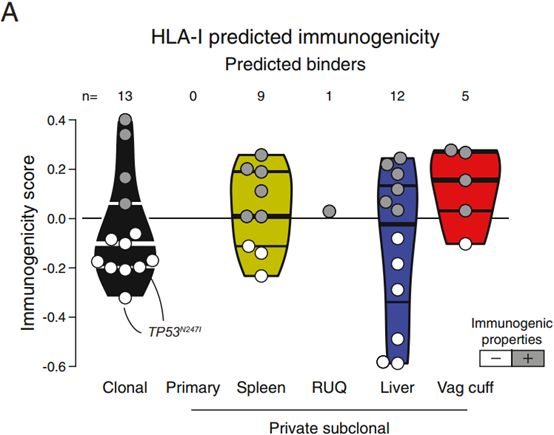
肿瘤抗原克隆性(Neoantigen Clonality)
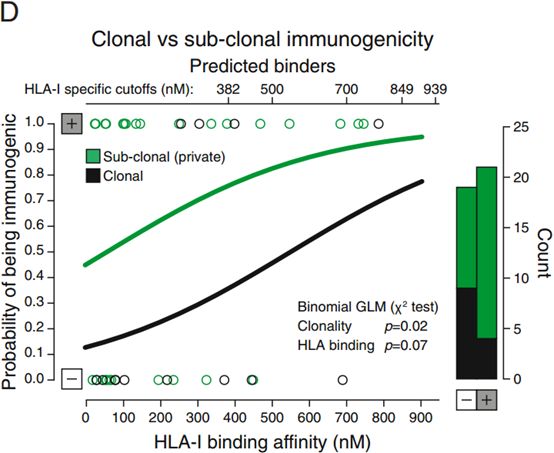
肿瘤抗原克隆性对肿瘤抗原免疫原性有影响。一般来说克隆突变具有更高的免疫原性,不过也有文章得出相反的结论。肿瘤抗原克隆性和肿瘤抗原免疫原性之间的关系表明肿瘤抗原发展过程的一个负向选择过程[3]。We observed that there was a significant effect of neoepitope clonality on the probability of a neoepitope having immunogenic properties, with clonal neoepitopes being predicted as less immunogenic (Figures S5A–S5D).[3]
肿瘤抗原与MHC复合物(pMHC)
可以分析在不同癌型之间pMHC的情况,可以比较其中的p也可以比较MHC出现频数。The number of pMHCs (neoantigen load) varied between immune subtypes (Figure 1C).[2]

T细胞受体库与肿瘤抗原(TCR-Neoantigen)
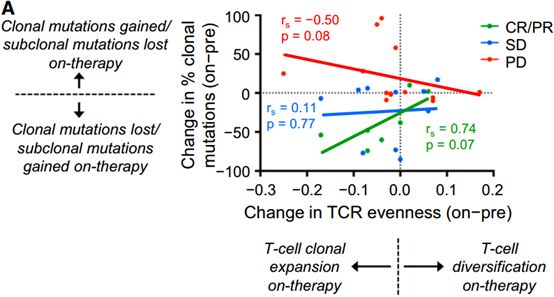
TCR能影响肿瘤抗原的发展,决定其的发展方向。这两者之间有着动态进化的关系[4]。Changes in T cell repertoire evenness were directly proportional to changes in the fraction of clonal mutations in patients with CR/PR and PD. Notably, there was a trend for a positive relationship in responders (p = 0.07) but a negative relationship in patients with PD (p = 0.08) (Figure 6A). [4]这点与第一点相似。
治疗前后对生成肿瘤抗原的突变与不能生成肿瘤抗原的突变(无意突变)的比值(the ratios of mutations that produce predicted neoantigens to those that do not)
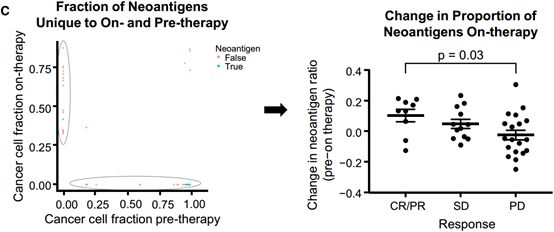
是否在临床上不能检测到的突变更倾向于生成肿瘤抗原。To investigate whether mutations that became undetectable on-therapy were more likely to be neoantigens or missense mutations than nonantigenic or synonymous mutations, the ratios of mutations that produce predicted neoantigens to those that do not were compared between mutations detected solely on-therapy and those detected solely pre-therapy.[4]
分析详情
Neoantigen Prediction
1:The localized peptides that tile across and contain the mutated amino acid substitution are identified and parsed into the neoantigen prediction pipeline. Each peptide is considered for HLA binding strength relative to its non-mutant (wildtype) counterpart.[1]
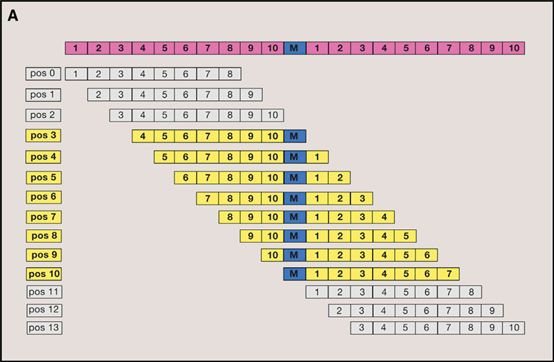
2:To predict neoepitopes, ‘‘wild-type’’ petide 17mers (for HLA-I) and 29mers (for HLA-II) with the affected amino acid in the middle for each missense mutation were retrieved from the GRCh37.74 human reference proteome. To generate ‘‘mutant’’ peptides, the affected amino acid was replaced in silico with the corresponding mutant amino acid.[3]
3:HLA typing with OptiType
HLA class I typing of samples (raw RNA-Seq from 8872 samples and aligned reads from 715 samples) was performed on the Seven Bridges Cancer Genomics Cloud using a Common Workflow Language (CWL) description of the OptiType tool. The aligned RNA-Seq samples were first converted to raw sequences using a CWL description of the Picard SamtoFastq tool. The reads from each raw RNA-Seq sample were first aligned to the HLA class I database using a CWL description of the yara aligner with its error rate parameter set to 3%. Next, the CWL description of OptiType was used to compute the HLA class I types for the sample. OptiType was run under its default parameters for RNA sequencing data using the GLPK linear programming solver and the CBC linear programming solver in samples where the GLPK solver failed. In order to validate the typing results from OptiType, we compared the HLA class I four-digit types obtained from the software PolySolver on TCGA Whole Exome Sequencing data samples (Shukla et al., 2015). For the 5222 patient cases shared by the two studies, approximately 90% of the typing calls were completely concordant for all HLA-A, HLA-B or HLA-C alleles, whereas completely discordant calls were found in less than 1.5% of cases for each of the genes.[2]
4:Neoantigen Prediction from SNVs
Potential neoantigenic peptides were identified using NetMHCpan v3.0 (Nielsen and Andreatta, 2016), based on HLA types derived from RNA-seq using OptiType as above. In brief, using the HLA calls from OptiType, for each sample, all pairs of MHC and minimal mutant peptide were input into NetMHCpan v3.0 using default settings. NetMHCpan will automatically extract all 8-11-mer peptides from a minimal peptide sequence and predict binding for each peptide-MHC pair. After computation, the results were parsed to only retain peptides which included the mutated position. Peptides containing amino acid mutations were identified as potential antigens on the basis of a predicted binding to autologous MHC (IC50 < 500 nM) and detectable gene expression meeting an empirically determined threshold of 1.6 transcripts-per-million (TPM). This threshold was selected in order to divide the bimodal distribution in the expression data.[2]
Specifically, somatic nonsynonymous coding single nucleotide variants were extracted from the MC3 variant file with the following filters: FILTER in ‘‘PASS,’’ ‘‘wga,’’ ‘‘native_wga_mix’’; NCALLERS > 1; barcode in whitelist where do_not_use = False; Variant_Classification = ‘‘Missense_Mutation’’; and Variant_Type = ‘‘SNP.’’ For each SNV, the Ensembl protein reference sequence was obtained, and the minimal peptide encompassing the mutation site plus 10 amino acids up and downstream of the mutation site was extracted (21 aa long peptide). If the mutation occurred within 10 amino acids of the N- or C-terminal end of the protein, all available sequence between the mutation and start/end of the protein was taken, resulting in a minimal peptide shorter than 21 aa. The variant position within the minimal peptide was recorded, and the mutation was applied to the minimal peptide, resulting in a mutant minimal peptide. Variation in sequencing coverage and tumor purity require careful consideration in order to mitigate the risk of impacting mutation calls and on pMHC, and prior to pMHC calling, sequencing data was subjected to rigorous harmonization efforts, performed by the PanCancer MC3 Consortium.[2]
5:Neoantigen Prediction from Indels
Somatic indel variants were extracted from the MC3 variant file with the following filters: FILTER in ‘‘PASS,’’ ‘‘wga,’’ ‘‘native_wga_mix’’ (with no combination with other tags); NCALLERS > 1; barcode in whitelist where do_not_use = False; Variant_Classification = ‘‘Frame_Shift_Ins,’’ ‘‘Frame_Shift_Del,’’ ‘‘In_Frame_Ins,’’ ‘‘In_Frame_Del,’’ ‘‘Missense_Mutation,’’ “Nonsense_Mutation’’; and Variant_Type = ‘‘INS,’’ ‘‘DEL.’’ For each Indel, the downstream protein sequence was obtained using VEP v87 (Ensembl Variant Effect Predictor) using default settings. Using 9-mer peptides extracted from VEP downstream protein sequences and the HLA calls from OptiType, for each sample, binding for each pair of mutant peptide-MHC were predicted using pVAC-Seq pipeline with NetMHCpan using default settings, of which an IC50 binding score threshold 500 nM was used to report the predicted binding epitopes as neoantigens.[2]
6: Prediction of candidate neoantigens from next-generation sequencing (NGS) data requires the implementation of several computational tasks
prediction of mutated peptides from whole-exome sequencing (WES), whole-genome sequencing (WGS) or RNA sequencing (RNA-seq) data from matched tumour–normal samples; selection of expressed peptides by integrating RNA-seq data of the tumour sample; human leukocyte antigen (HLA) typing from WES, WGS or RNA-seq data of the tumour sample; and prediction of peptide–MHC binding for specific HLA alleles.[5]
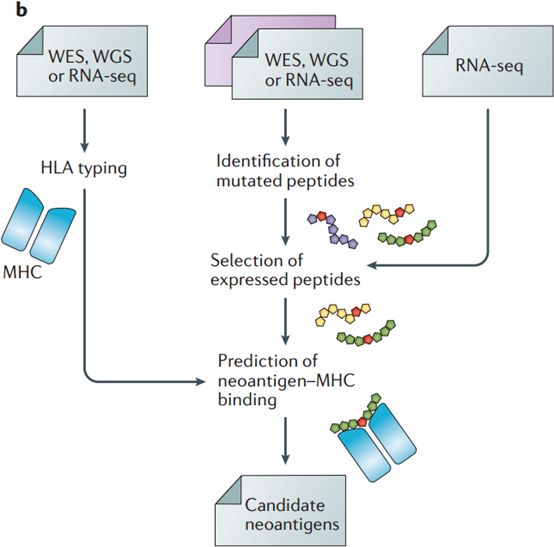
7: High-resolution HLA typing was performed computationally using SOAP-HLA from exome sequencing (exome-seq) data.
Each nonsynonymous SNV was translated into a 17-mer peptide sequence, centered on the mutated amino acid. Adjacent SNVs (such as those induced by UV irradiation) were first corrected for using MAC. Subsequently, the 17-mer was then used to create 9-mers via a sliding window approach for determination of MHC class I binding. NetMHC was used to determine the binding strength of mutated peptides to patient-specific HLA alleles. All peptides with a rank < 2% were considered for further analysis. If one mutation generated multiple 9-mer peptides that bound to patient-specific HLA alleles, it was only counted as one neoantigen (i.e., one mutation could only generate a single predicted neoantigen). We considered neopeptides as the total number of predicted 9-mers that bound to patient-specific HLA alleles (i.e., one mutation can generate multiple neopeptides).[4]
To test whether the observed number of neoantigens was different from the expected count in each on-therapy tumor sample, we empirically calculated the following baseline rates pre-therapy:
1) The average number of missense mutations per silent mutation across tumors.
2) The number of neoantigens per missense mutation. Of note, since computational tools that predict neoantigens are dependent on patient-specific HLA class I alleles and some HLA-I alleles can bind greater numbers of peptides than others, we calculated this rate specific to the patient, rather than an average across all patients.
Using these two baseline rates, we used the number of silent mutations in each on-therapy tumor sample to calculate the expected number of neoantigens. Hence, the expected number of neoantigens in an on-therapy tumor sample is equal to the number of silent mutations in an on-therapy tumor sample multiplied by the number of missense mutations per silent mutation pre-therapy, multiplied by the number of neoantigens per missense mutation in the patient’s tumor sample pre-therapy. The chi-square test was used to determine whether the observed number was different from the expected value. All the calculations were done using R statistical software.[4]
Neoepitope depletion analysis
To analyze neoepitope depletion across the different samples, we followed the method developed by Rooney and colleagues using only expressed mutations. Commonly mutated genes were not included as indicated (Rooney et al., 2015).[3] (具体可见原文)
[1] LIU X S, MARDIS E R. Applications of Immunogenomics to Cancer [J]. Cell, 2017, 168(4): 600-12.
[2] THORSSON V, GIBBS D L, BROWN S D, et al. The Immune Landscape of Cancer [J]. Immunity, 2018, 48(4): 812-30 e14.
[3] JIMENEZ-SANCHEZ A, MEMON D, POURPE S, et al. Heterogeneous Tumor-Immune Microenvironments among Differentially Growing Metastases in an Ovarian Cancer Patient [J]. Cell, 2017, 170(5): 927-38 e20.
[4] RIAZ N, HAVEL J J, MAKAROV V, et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab [J]. Cell, 2017, 171(4): 934-49 e15.
[5] HACKL H, CHAROENTONG P, FINOTELLO F, et al. Computational genomics tools for dissecting tumour-immune cell interactions [J]. Nature reviews Genetics, 2016, 17(8): 441-58.