测序上游分析系列:
mRNA-seq转录组二代测序从raw reads到表达矩阵:上中游分析pipeline
miRNA-seq小RNA高通量测序pipeline:从raw reads,鉴定已知miRNA-预测新miRNA,到表达矩阵【一】
miRNA-seq小RNA高通量测序pipeline:从raw reads,鉴定已知miRNA-预测新miRNA,到表达矩阵【二】
其他文章系列:ggplot2作图篇:
(1)基于ggplot2的RNA-seq转录组可视化:总述和分文目录
(2)测序结果概览:基因表达量rank瀑布图,高密度表达相关性散点图,注释特定基因及errorbar的表达相关性散点图绘制
(3)单/多个基因在组间同时展示的多种选择:分组小提琴图、分组/分面柱状图、单基因蜂群点图拼图的绘制
(4)配对样本基因表达趋势:优化前后的散点连线图+拼图绘制
常见的TCGA数据挖掘办法之一,是通过差异基因分析获得差异表达基因,然后从中筛选出部分表达水平与患者生存相关联的候选基因,对它们的表达水平进行多因素cox回归构建风险模型,评估风险模型的预测能力(ROC曲线),并用Kaplan-Meier生存分析评估模型的风险评分是否有意义。
本例仍使用之前教程选择的的TCGA-LUSC白色人种肺鳞癌表达谱数据。
TCGA数据下载整合操作见前文:从TCGA数据库下载并整合清洗高通量肿瘤表达谱-临床性状数据
DESeq2差异分析见前文:TCGA数据整合后进行DESeq2差异表达分析和基于R的多种可视化
本文使用的数据为下载的HTSeq-counts数据,已经通过了整合和删拾数据的清洗,并通过DESeq2完成了差异分析。本文用到的变量和对象为:
(1) dds_DE: the object generated by DESeq2. It contains the results of differential expression such as symbols of DE-genes and normalized counts.
(2) condition_table_cancer: the data frame containing the TCGA_IDs, overall_survival and vital status of cancer patients.
需要的R包:DESeq2包(转录组差异分析),survival包(cox回归),survminer包(Kaplan-Meier Plot可视化),dplyr包(字符串处理),glmnet包(Lasso回归),ggplot2包(数据可视化),GGally包(绘制相关性矩阵图),rms包(计算VIF方差膨胀因子),survivalROC包(绘制time dependent ROC曲线),plotROC包(绘制ROC曲线)。
install.packages(c('DESeq2', 'survival', 'survminer', 'dplyr', 'glmnet', 'ggplot2', 'GGally', 'rms', 'survivalROC', 'plotROC'))
library('DESeq2')
library('survival')
library('survminer')
library('dplyr')
library('glmnet')
library('ggplot2')
library('GGally')
library('rms')
library('survivalROC')
library('plotROC')
1. 获取差异表达基因,以及差异基因的标准化counts矩阵。
我们假设现在DESeq2已经做好差异分析,获得了dds_DE object!
使用results函数获取cancer组相对normal组差异表达p值<0.05的基因和相关信息。随后选取符合|log2FoldChange|差异大于2(|FoldChange|大于4)和FDR<0.01的差异基因子集。resSigAll对象的rownames记录了差异基因名信息。
随后获取基因的normalized counts matrix并做vst转换,仅提取差异基因的normalized counts进行后续分析。
res_DE <- results(dds_DE, alpha=0.05,contrast=c("sample_type","cancer","normal"))
resOrdered <- res_DE[order(res_DE$padj),]
resSig <- subset(resOrdered,padj < 0.01)
resSigAll <- subset(resSig, (log2FoldChange < (-2)|log2FoldChange > 2))
rld <- vst(dds_DE, blind = T)
expr_norm <- assay(rld)
DESeq_norm_vst_for_survival <- expr_norm[resSigAll@rownames,]
2. 构建回归分析所需的信息表。
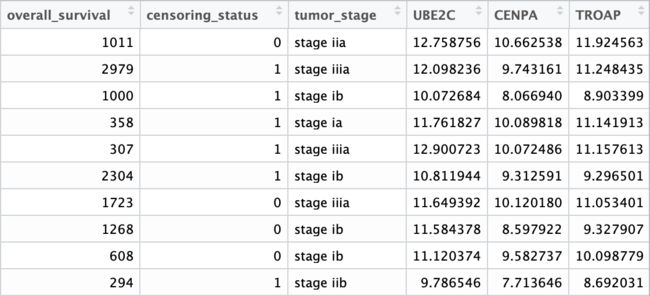
采用Survival R包对下载数据进行回归分析,需要构建一个至少包含样本生存时间、删失状态的data frame。本例中,在数据整合建立condition_table时已经获得了一个cancer patients的子集condition_table_cancer,直接使用它建立信息表,并合并潜在预后预测因子:候选基因的normalized counts(t()转换至行为样本,列为基因)。
注:survival包认定0为censored(无事件发生/删失),1为diseased(有结局事件发生)。所以在本例中'Alive'转换为0,'Dead'转换为1。
此外有基因名中带有R不被认可的dash '-',所以将data frame中colnames的 '-' 转换为下划线 '_'。
library('dplyr')
survival_cancer <- condition_table_cancer
#Alive=0, Dead=1
survival_cancer$censoring_status <- gsub(survival_cancer$censoring_status, pattern = 'Alive', replacement = '0') %>%
gsub(pattern = 'Dead', replacement = '1') %>% as.numeric()
rownames(survival_cancer) <- survival_cancer$TCGA_IDs
survival_cancer <- cbind(survival_cancer, t(DESeq_norm_vst_for_survival)[survival_cancer$TCGA_IDs,])
#formula function in the next step will not recognize gene names like 'NKX1-2'. We have to change '-' to '_'.
colnames(survival_cancer) <- gsub(colnames(survival_cancer), pattern = '-', replacement = '_')
3. 对差异基因进行批量单因素COX回归。
很多研究使用了先批量单因素COX回归选出更少的候选基因,随后进行多因素COX回归建模的策略。虽然这个策略由于忽略了变量间的相互关系而非常有争议,但是这里暂且不论策略对错,我们先用代码尝试实现批量单因素COX回归。
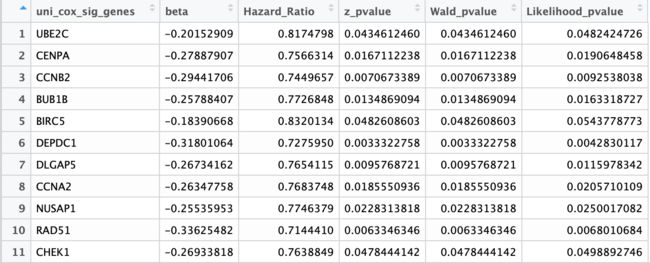
单因素COX回归建模依赖于survival包coxph()函数,PH检验依赖于cox.zph()函数。这里的思路是自建一个函数,然后输入需批量操作的基因名vector,和已经加入这些基因normalized counts的survival_cancer信息表。通过R特有的向量化操作lappy同时进行批量计算各基因的风险比,并检验变量是否符合cox等比例风险模型的前提PH假设,得到符合z值p值<0.05和符合PH假定(cox.zph检验p值>0.05)的基因。最终输出含有gene名,单因素cox的β值,危险比HR,z值的p值,Wald检验和似然比检验p值的data frame。
注:gene名需要时刻注意,将'-'转换为'_'才能与survival_cancer匹配。函数中已镶嵌这个转换。
#filter potential useful sig genes using univariate cox regression.
uni_cox_in_bulk <- function(gene_list, survival_info_df){
library('survival')
gene_list <- gsub(gene_list, pattern = '-', replacement = '_')
uni_cox <- function(single_gene){
formula <- as.formula(paste0('Surv(overall_survival, censoring_status)~', single_gene))
surv_uni_cox <- summary(coxph(formula, data = survival_cancer))
ph_hypothesis_p <- cox.zph(coxph(formula, data = survival_cancer))$table[1,3]
if (surv_uni_cox$coefficients[,5]<0.05 & ph_hypothesis_p>0.05){ #get the pvalue
single_cox_report <- data.frame('uni_cox_sig_genes'=single_gene,
'beta'=surv_uni_cox$coefficients[,1],
'Hazard_Ratio'=exp(surv_uni_cox$coefficients[,1]),
'z_pvalue'=surv_uni_cox$coefficients[,5],
'Wald_pvalue'=as.numeric(surv_uni_cox$waldtest[3]),
'Likelihood_pvalue'=as.numeric(surv_uni_cox$logtest[3]))
single_cox_report
}
}
uni_cox_list <- lapply(gene_list, uni_cox)
do.call(rbind, uni_cox_list)
}
uni_cox_df <- uni_cox_in_bulk(gene_list = resSigAll@rownames, survival_info_df = survival_cancer)
最终得到279个有意义的基因。输出的data frame如图:
Optional:
以上的代码嵌套了两次自定义函数,虽然不是最优化的做法,但较好地利用了R的向量化特性。如下代码的目的是等价的,也是批量单因素cox回归,但放弃了向量化,在R中使用了for循环,运行速度明显慢于上方代码。
uni_cox_sig_genes <- c()
beta_co <- c()
HR <- c()
z_p <- c()
Wald_p <- c()
Likelihood_p <- c()
for (candidate_gene in gsub(resSigAll@rownames, pattern = '-', replacement = '_')){
formula <- as.formula(paste0('Surv(overall_survival, censoring_status)~', candidate_gene))
surv_uni_cox <- summary(coxph(formula, data = survival_cancer))
ph_hypothesis_p <- cox.zph(coxph(formula, data = survival_cancer))$table[1,3]
if (surv_uni_cox$coefficients[,5]<0.05 & ph_hypothesis_p>0.05){ #get the pvalue
uni_cox_sig_genes <- append(uni_cox_sig_genes, candidate_gene)
beta_co <- append(beta_co, surv_uni_cox$coefficients[,1])
HR <- append(HR, exp(surv_uni_cox$coefficients[,1]))
z_p <- append(z_p, surv_uni_cox$coefficients[,5])
Wald_p <- append(Wald_p, as.numeric(surv_uni_cox$waldtest[3]))
Likelihood_p <- append(Likelihood_p, as.numeric(surv_uni_cox$logtest[3]))
}
}
uni_cox_df <- data.frame('uni_cox_sig_genes'=uni_cox_sig_genes, 'beta'=beta_co, 'Hazard_Ratio'=HR, 'z_pvalue'=z_p, 'Wald_pvalue'=Wald_p, 'Likelihood_pvalue'=Likelihood_p)
4. Lasso回归筛选具有代表性的变量。
单因素回归筛选出的变量仍旧较多,我们尝试用Lasso回归获得较少有意义的变量。Lasso回归的前提是变量数(这里是差异基因,n=2726)>样本数(这里是cancer patients数,n=344),回归的目的是使deviance最小化,结果是将不重要变量的系数压缩为0,仅保留较少的重要变量系数不为0。
Lasso回归依赖glmnet包。我个人下载这个包的时候遇到了R版本不匹配的问题,如果有朋友也遇到无法install.packages的问题,可以右转github下载,或网页版CRAN手动下载安装,这里不多说。
注:glmnet需要输入x的格式为矩阵(matrix),y的格式为double,否则会报错。
#about glmnet: x should be in format of matrix, and time&status in y should be in double format.
x <- as.matrix(survival_cancer[,gsub(resSigAll@rownames, pattern = '-', replacement = '_')])
y <- survival_cancer[,c('overall_survival', 'censoring_status')]
names(y) <- c('time', 'status')
y$time <- as.double(y$time)
y$status <- as.double(y$status)
y <- as.matrix(survival::Surv(y$time, y$status))
lasso_fit <- cv.glmnet(x, y, family='cox', type.measure = 'deviance')
coefficient <- coef(lasso_fit, s=lasso_fit$lambda.min)
Active.Index <- which(as.numeric(coefficient) != 0)
active.coefficients <- as.numeric(coefficient)[Active.Index]
sig_gene_multi_cox <- rownames(coefficient)[Active.Index]
这一次我们仅仅获得了4个candidates:

5. 利用筛选出来的少数candidates建立多因素COX回归模型。
话不多说,依旧用survival包的cox.ph()建模,用coxzph()检验每个因子的PH假设。然后计算回归模型中每个因子的VIF和相关系数,判断因素间可能存在的共线性。通过PH假设和共线性检验的变量再进行第二次建模,随后绘制COX回归森林图。
#perform the multi-variates cox regression using qualified genes.
formula_for_multivariate <- as.formula(paste0('Surv(overall_survival, censoring_status)~', paste(sig_gene_multi_cox, sep = '', collapse = '+')))
multi_variate_cox <- coxph(formula_for_multivariate, data = survival_cancer)
#check if variances are supported by PH hypothesis.
ph_hypo_multi <- cox.zph(multi_variate_cox)
#The last row of the table records the test results on the GLOBAL model. Delete it.
ph_hypo_table <- ph_hypo_multi$table[-nrow(ph_hypo_multi$table),]
#Remove variances not supported by ph hypothesis and perform the 2nd regression.
formula_for_multivariate <- as.formula(paste0('Surv(overall_survival, censoring_status)~', paste(rownames(ph_hypo_table)[ph_hypo_table[,3]>0.05], sep = '', collapse = '+')))
multi_variate_cox_2 <- coxph(formula_for_multivariate, data = survival_cancer)
到这里我们可以发现4个candidates中有1个未通过coxzph()的检验,只有3个基因纳入了第二次的建模。

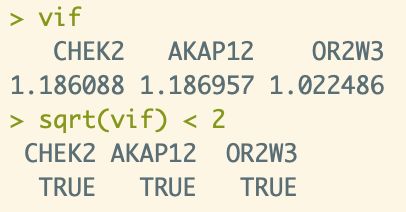
关于检测变量间的共线性,需要综合考虑:变量间的相关系数<0.5和vif平方根<2均被提出是可行的检验方法。
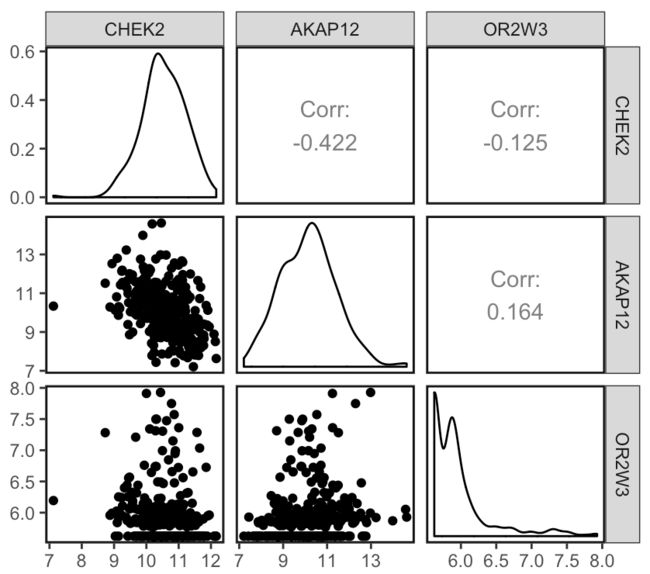
我们进行相关系数,vif的计算,并进行相关性矩阵的可视化。
#check the co-linearity between samples.
correlation <- cor(survival_cancer[,rownames(ph_hypo_table)[ph_hypo_table[,3]>0.05]], method = 'pearson')
library('GGally')
ggpairs(survival_cancer[,rownames(ph_hypo_table)[ph_hypo_table[,3]>0.05]],
axisLabels = 'show')+
theme_bw()+
theme(panel.background = element_rect(colour = 'black', size=1, fill = 'white'),
panel.grid = element_blank())
library('rms')
vif <- rms::vif(multi_variate_cox_2)
#Some people said if the square root of VIF >2, they might be co-linear.
sqrt(vif) < 2
可以得到结果:变量vif均未达到共线性的程度。不需进一步剔除。
相关性矩阵可视化结果如下:
接下来用森林图可视化这个模型。
ggforest(model = multi_variate_cox_2, data = survival_cancer, main = 'Hazard ratios of candidate genes', fontsize = 1)
可视化结果如下,记录了模型中各因子的危险比HR,置信区间,模型的AIC值,concordance index。结果说明模型中OR2W3为独立的显著危险因素,CHEK2为独立的显著保护因素,且整个cox模型也具有显著意义:

6. 多方面评价cox模型的预测能力。
(1)Concordance index:
模型的一致性系数反映了其预测能力。C-index=0.5则完全随机,=1为预测与实际完全一致。>=0.9为高,0.7-0.9间为中等,0.5-0.7间为低。
C_index <- multi_variate_cox_2$concordance['concordance']
if(C_index >= 0.9){
print('High accuracy')
}else{
if(C_index < 0.9 & C_index >= 0.7){
print('Medium accuracy')
}else{
print('Low accuracy')
}
}
Anyway, just have fun...
(2)time-dependent ROC curve:
ROC曲线用于反映预测模型的准确性和精确性,AUC曲线下面积越大,ROC曲线转折点越靠近左上角,说明预测能力越好。一般认为AUC>=0.7可以够看。
对于COX回归,可以使用time-dependent ROC曲线检测模型预测特定时间预后的能力。
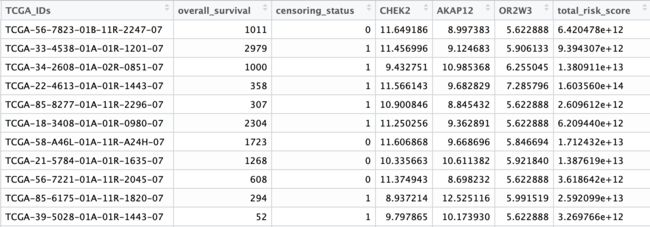
COX建模的意义在于用模型计算出的risk score预测病人的生存状态。首先用模型得到的系数计算每个样本的risk score(gene1β1+gene2β2+gene3*β3):
依旧是自建函数,输入survival_cancer信息表,candidate genes vector和多因素cox回归对象,输出包含每个样本risk score的risk_score_table.
#calculate the risk score of each sample.
riskscore <- function(survival_cancer_df, candidate_genes_for_cox, cox_report) {
library('dplyr')
risk_score_table <- survival_cancer_df[,candidate_genes_for_cox]
for(each_sig_gene in colnames(risk_score_table)){
risk_score_table$each_sig_gene <- risk_score_table[,each_sig_gene]*(summary(cox_report)$coefficients[each_sig_gene,1])
}
risk_score_table <- cbind(risk_score_table, 'total_risk_score'=exp(rowSums(risk_score_table))) %>%
cbind(survival_cancer_df[,c('TCGA_IDs','overall_survival','censoring_status')])
risk_score_table <- risk_score_table[,c('TCGA_IDs','overall_survival','censoring_status', candidate_genes_for_cox, 'total_risk_score')]
risk_score_table
}
candidate_genes_for_cox2 <- c(rownames(ph_hypo_table)[ph_hypo_table[,3]>0.05])
risk_score_table_multi_cox2 <- riskscore(survival_cancer, candidate_genes_for_cox2, multi_variate_cox_2)
这里我们自建函数,批量做3-5年多个时间点的ROC曲线,比较出AUC最大的时点。
multi_ROC <- function(time_vector, risk_score_table){
library('survivalROC')
single_ROC <- function(single_time){
for_ROC <- survivalROC(Stime = risk_score_table$overall_survival,
status = risk_score_table$censoring_status,
marker = risk_score_table$total_risk_score,
predict.time = single_time, method = 'KM')
data.frame('True_positive'=for_ROC$TP, 'False_positive'=for_ROC$FP,
'Cut_values'=for_ROC$cut.values, 'Time_point'=rep(single_time, length(for_ROC$TP)),
'AUC'=rep(for_ROC$AUC, length(for_ROC$TP)))
}
multi_ROC_list <- lapply(time_vector, single_ROC)
do.call(rbind, multi_ROC_list)
}
#We evaluate 11 AUCs between 3-5 years.
for_multi_ROC <- multi_ROC(time_vector = c(365*seq(3,5,0.2)), risk_score_table = risk_score_table_multi_cox2)
输出的for_multi_ROC dataframe中含有每个时间点的True positive和False positive随不同cut off值变化的趋势。

不同时间点ROC曲线的可视化:
#visualization of the ROC curves of multiple time points.
pROC<-ggplot(for_multi_ROC, aes(x = False_positive, y = True_positive, label = Cut_values, color = Time_point)) +
geom_roc(labels = F, stat = 'identity', n.cuts = 0) +
geom_abline(slope = 1, intercept = 0, color = 'red', linetype = 2)+
theme_bw()+
theme(panel.background = element_rect(colour = 'black', size=1, fill = 'white'),
panel.grid = element_blank())+
annotate("text",x = 0.75, y = 0.15,
label = paste("AUC max = ", round(AUC_max, 2), '\n', 'AUC max time = ', AUC_max_time, ' days', sep = ''))
pROC
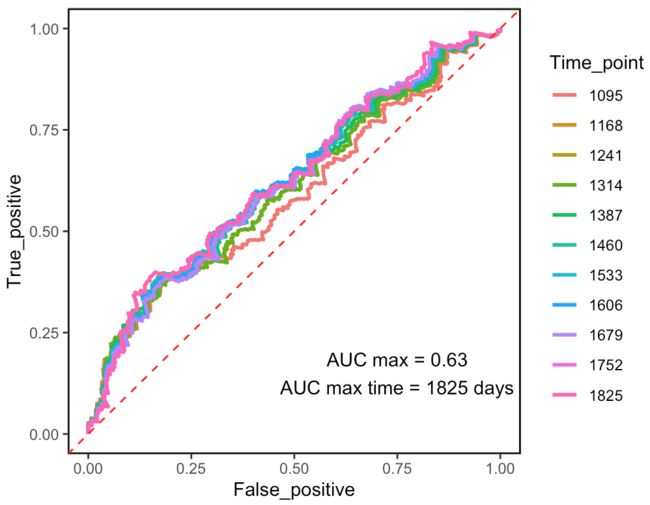
AUC max为0.63,可见其实预测能力都很差- -菜鸡互啄罢了。只是案例操作,无妨。
(3) Kaplan-Meier生存分析。
Kaplan-Meier分析用于比较两组之间的生存状态。所以我们需要把计算出来的risk_score按照一个截点分为高/低风险组。
ROC曲线的另外一个功能就是可以用于risk score分组最佳截点的选择。
首先选取AUC最大的时间点,使用这个时间点的ROC曲线进行Kaplan-Meier分析。
然后截取ROC曲线的转折点,也就是真阳性和假阳性差值最大的点,作为risk score的cut-off值,高于这个值为高风险组,低于这个值为低风险组,并把信息并入risk_score_table中。
AUC_max <- max(for_multi_ROC$AUC)
#maybe AUCs are identical in different time points. So select the last time point indicating longer survival.
AUC_max_time <- for_multi_ROC$Time_point[which(for_multi_ROC$AUC == AUC_max)]
AUC_max_time <- AUC_max_time[!duplicated(AUC_max_time)]
AUC_max_time <- AUC_max_time[length(AUC_max_time)]
for_multi_ROC$Time_point <- as.factor(for_multi_ROC$Time_point)
#find the optimal cutoff value within the ROC curve of the optimal time point.
optimal_time_ROC_df <- for_multi_ROC[which(for_multi_ROC$Time_point == AUC_max_time),]
cut.off <- optimal_time_ROC_df$Cut_values[which.max(optimal_time_ROC_df$True_positive-optimal_time_ROC_df$False_positive)]
high_low <- (risk_score_table_multi_cox2$total_risk_score > cut.off)
high_low[high_low == TRUE] <- 'high'
high_low[high_low == FALSE] <- 'low'
risk_score_table_multi_cox2 <- cbind(risk_score_table_multi_cox2, high_low)
此时,已经将risk score这个连续变量转变为了'high'和'low'的二分类变量。使用survival包建立KM分析对象,然后使用survminer包可视化作KM-plot并进行Log-rank分析。
注:为了最佳的可视化效果,根据之前获得的最佳预测时间(本例为1825天=5年),将Overall_survival超过预测时间的样本编辑一下,将生存时间改为1825天,生存状态改为0。(可理解为观察终点为5年,这些样本在这个时点成功完成观察而未发生事件)
#KM_plot generation.
library('survminer')
#first edit the status of patients with OS > AUC max time. (censoring status=0 (Alive), OS=365*5 days)
risk_score_table_multi_cox2$censoring_status[which(risk_score_table_multi_cox2$overall_survival > AUC_max_time)] <- 0
risk_score_table_multi_cox2$overall_survival[which(risk_score_table_multi_cox2$overall_survival > AUC_max_time)] <- AUC_max_time
fit_km <- survfit(Surv(overall_survival, censoring_status) ~high_low, data = risk_score_table_multi_cox2)
ggsurvplot(fit_km, conf.int = F,pval = T,legend.title="total risk score",
legend.labs=c(paste0('>',as.character(round(cut.off,2))), paste0('<=',as.character(round(cut.off,2)))), risk.table = T,
palette = c('red','blue'), surv.median.line = 'hv')
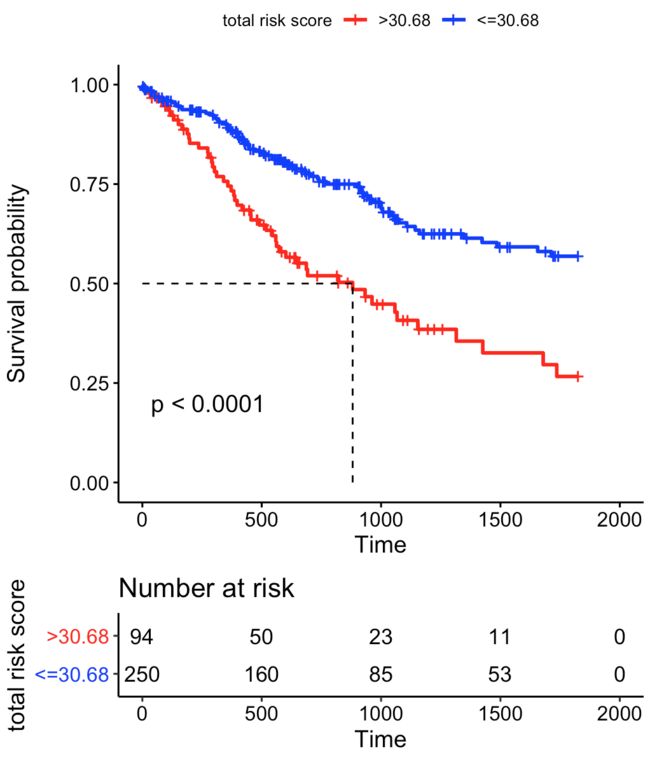
至此时,一整套生存分析就完成了!
写这么多,我太难了...