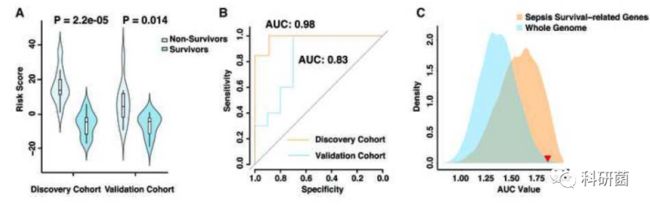
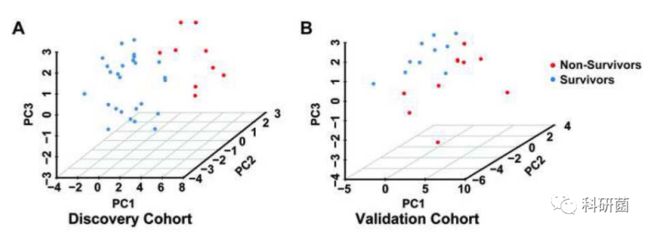
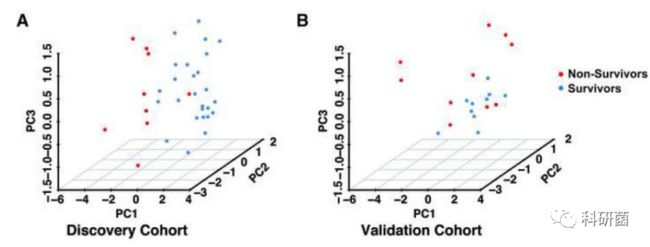
- 2018年11月24日亲子日记第189篇
涵飞儿_dcfa
周六孩子最忙碌的一天!早上让孩子多睡了会,还好没耽误学舞蹈。下午2点学钢琴,老师说弹的还可以,学新曲子也挺快。心里听了还挺舒服。回来休息了一会儿又赶去写字,到了6点接上孩子又去学乐理知识。以前周六周日是我自己在家接送宝贝!这周宝贝奶奶没回老家,看到孩子一天忙个不停,开始念叨着孩子比上学还累。看来是老人心疼孩子了!晚上给孩子多加了一个菜,我也跟着沾光了哈哈!还好晚上吃饭奶奶没有说别的!我也感觉孩子一
- 2018-10-24丨微日记027
Jonathanchoi
今天分享一些小碎片:有道云笔记里头有一个扫描文档的功能,可是它只能自动识别,却不能给用户编辑的机会,可谓是“拍得到就是你的,拍不到就拜拜”,而扫描全能王则提供了识别错误后可编辑的功能。开完组会路过包道的时候,发现它提供了顾客到店开柜取餐的功能,为想吃到美食却赶路程赶时间的人们提供了多种选择性。这种饮食界的丰巢快递柜,个人我觉得很实用。捷登都会的洗手间设置让人不太习惯,三层男厕二层是女厕,经常让人白
- 2021.12.15 晴 星期三 孙贞正妈妈亲子日记第
秋枫_d581
今天早上起床,因为爸爸早上生了炉子,所以早上起来不会特别的冷,但是外面特别的冷,收拾好了书包,就去上学了,外面实在是特别特别的冷,因为车上已经喷上防冻玻璃水了,还是会起霜,之后就去上学了,早读上的是数学,数学讲了昨天晚上写的作业,但是没有讲完就下课了,第一节课上的也是数学,讲了早读没有讲完昨天晚上作业,第二节课上的是英语,上完英语之后,就去跑操了,但是我一跑就腿疼,所以没有去跑,之后就去上第三节课
- c++内存管理与模板初阶
Slowstep_
c++c语言数据结构
文章目录虚拟进程地址空间区域new和deletenew的失败机制new/delete原理重载operatornew和operatordeletenew[]/delete[]定位newnew多维数组模板虚拟进程地址空间c/c++的一个程序在内存区域中的划分:由低地址到高地址依次是:代码区->已初始化全局数据区->未初始化全局数据区->堆区->栈区。其中堆区和栈区是相对而生的,堆区和栈区之间有很大的镂
- 20220415星期五晴
像风一样飞驰
早上右边打屁股针+手部踩车20分钟+右肩薰蒸30分钟+腰部及屁股针炙30分钟,下午右手前臂电疗20分钟+经颅磁刺激议20分钟+康复训练60分钟。今天三个病人出院44床二个台阶看成一个台阶踩空摔倒右手肩部骨拆有做过手术,一开始手抬不高现在可以抬过头顶了,64床的一个阿姨原本不会走路出院时走路已走的很稳很快,69床的丁莲英腰部摔伤已经好了,就是出院前一直低烧打了两天的青霉素才有所下降,另外验血发现缺钾
- 咏怀八十二首·其三十二
紫雁东来
[魏晋]阮籍朝阳不再盛,白日忽西幽。去此若俯仰,如何似九秋。人生若尘露,天道邈悠悠。齐景升丘山,涕泗纷交流。孔圣临长川,惜逝忽若浮。去者余不及,来者吾不留。愿登太华山,上与松子游。渔父知世患,乘流泛轻舟。译文:朝阳终究不能始终灿烂下去,随着白日西落,天色也很快变得幽暗。去这里时间短暂,如何能像很长时间。人生就如同尘土就如同朝露,很快地就会消失,而“悠悠”天道却永恒长久。在齐景公登牛山,见山川之美,
- 金牛山人
口水说电影
金牛山人1984年发现,遗址在辽宁营口县西金牛山的洞穴里。化石有较完整的头骨1个,还有脊椎骨、肋骨、腕骨、掌骨、趾骨、髋骨、尺骨、指骨、跗骨等比较罕见的猿人骨骼化石50余件。金牛山人距今28万年左右。金牛山人1984年发现,遗址在辽宁营口县西金牛山的洞穴里。化石有较完整的头骨1个,还有脊椎骨、肋骨、腕骨、掌骨、趾骨、髋骨、尺骨、指骨、跗骨等比较罕见的猿人骨骼化石50余件。金牛山人距今28万年左右。
- 葱油桃酥饼
清凌凌的遂
葱油桃酥饼:材料:鸡蛋一个,猪油250克,低筋粉500克,葱160克,糖60克,盐10克,泡打粉5克,食用小苏打5克。步骤:1,先将鸡蛋、糖和猪油2分钟速度3打发。2,加入低筋粉、盐和葱速度1到6混合一分钟。3:取出面团稍微揉一下分成30克左右的剂子,搓圆然后压成圆形,然后用拇指按压中心位置形成一个小圆坑即可。4:烤箱200度12分钟图片发自App
- 《白鹿原》读后感2
fjutlxl
第二章则是围绕着白鹿原这个地名中的两字展开的。白鹿作为原上劳动人民的一种精神信仰,其所发挥的潜在作用是无法估量的。白嘉轩听从冷先生的建议出门找个先生来看看家宅风水,因途中一次解手而得显现于鹿子霖慢坡地上的白鹿精灵,原本不知是怪物或者祥瑞的他,请教于堪为五总之龟的姐夫朱先生,朱先生让嘉轩画了所见之物,思索片刻后得出这是白鹿的结论,白鹿对于白鹿原上的人来说,就是祥瑞。可是为了得到上天所赐予的祥物,嘉轩
- 迪奥官网口红优惠码,乐分享注册邀请码63天陪跑计划是什么?
优惠券高省
迪奥口红优惠券从哪里买便宜强烈推荐,氧惠APP必须要邀请码才可以注册吗?查看更多关于迪奥口红优惠券从哪里买便宜强烈推荐,氧惠APP必须要邀请码才可以注册吗?的文章刚才我说了,一共9层,第九层的人买第一单东西的佣金第八层的人获得,买第二次东西的时候第七层的人获得佣金......同样的你第一层5人的第十笔推广奖励给你,也是9笔一循环。来氧惠,给你一个赚钱、省钱的机会没错,你1层5个人的第一笔推广奖励给
- 《论语•公冶长篇》|| 明见己错而自省
唐古拉的呼唤
5.27子曰:“已矣乎!吾未见能见其过而内自讼者也。”译意:孔子说:“算了吧,我还没有看见过能够看到自己的错误便能自我责备的人。”人非圣贤,孰能无过!人们都明白这个道理,然而自古以来,人们往往是对别人的错误能看得明白,对自己的错误却常常视而不见,或者明知自己确实有错,却找各种借口推诿避脱。孔子对此也很是感叹,说他根本没见过能够主动发现自己错误并且坦然承认,并自我反省的人。面对自己的错误时人常有两种
- 【实操】信息安全工程师系列-第22关 网站安全需求分析与安全保护工程
披荆斩棘的GG
安全
【实操】信息安全工程师系列-第22关网站安全需求分析与安全保护工程********永远不要信任用户输入。—安全编程格言一、网站安全基础概念与威胁分(一)核心定义**网站安全目标:**保障机密性(数据不泄露)、完整性(数据不被篡改)、可用性(服务不中断)和可控性(管理可控制)。**技术架构:**基于B/S架构,涉及网络通信、操作系统、数据库、Web服务器(如Apache、IIS)、Web应用及相关协
- 2021年,我在百家号里的美食碎碎念之二百五十五
暖暖的柠檬树
家里有现成的手擀面条,周末没有休息,下班回家比较累可,就没去菜市场买菜。用家里现有的食材做完热汤面。一颗西红柿炒成浓汁,加一勺肉臊子,白菜叶子,加入热水,水滚开后把面条直接煮到西红柿汤中,煮熟后,加入小青菜和合适的调味料,一起搅拌均匀就可以出锅了。热腾腾的一碗汤面,营养也不错,吃起来特别舒服。一碗简单的热汤面里藏着我的幸福和快乐!(以上美食碎碎念记录于2021年9月)
- 2023-06-21 最怀念
南方有佳木
今年的我,刚满四十。或许是生活得比较平顺、安稳,四十年来,我并没有觉得时光的无情流逝。增加的除了数字外,在我的心里,我依然还是那个27.8岁的我。但是,流逝的时光却永远带走了我的外公外婆。如果问我最想念的是谁?我想说最想念的是我的外公外婆,最怀念的时光是小时候在乡下和外公外婆一起生活的每一个假期。现在拟声造景的风吹麦浪,大雁南飞,瓜果飘香,在那个童年时光中,我都一一感受过。我很怀念那时的时光,我也
- 2022-05-21
人生_d8b6
中原焦点团队33期学员坚持分享第151天,2022.05.21约练43次,观4次,来39观察员我最近情绪不太稳定,如何稳定,情绪的背后是什么,在乎的点是什么?自己缺乏这些?怎么样解决自己的缺乏?犹豫的话将方案进行排序。
- 罗翔《刑法学讲义》精选摘录(下)
润苼
侵犯著作权罪44、基本刑处3年以下有期徒刑或者拘役,并处或者单处罚金,违法所得数额巨大或者有其他特别严重情节的,处3年以上10年以下有期徒刑,并处罚金。追诉标准是违法所得数额在3万元以上,或非法经营数额在5万元以上的。45、非法经营罪,基本刑处5年以下有期徒刑或者拘役,并处或者单处违法所得一倍以上五倍以下罚金,情节特别严重的,处5年以上有期徒刑,并处违法所得一倍以上五倍以下罚金或者没收财产。金融犯
- 2023-7-29晨间日记
木可柯98
今天是什么日子起床:9.00就寝:凌晨4点天气:台风天心情:焦虑任务清单昨日完成的任务,最重要的三件事:锻炼(羽毛球、撸铁)、看书、月复盘改进:1.在疯狂加班的这几个月里,终于找到时间锻炼了;2.在这高强度的工作中,你在不断地调整自己的工作方式和用几乎所有时间去按部就班处理琐碎事情,可能更多时候可以有选择性地做有意义地做事情,不一定要全盘接收!3.在更多做抉择的时候,会犹豫,不果断。4.在做紧急重
- 农业低息贷款如何影响你, 一系列农业金融优惠政策来袭
教你为人处事
江西的“财政惠农信贷通”是此类模式的典型代表。2014年,江西省、市、县三级财政筹集引导资金15亿元存入合作银行作为风险补偿金,合作银行按不低于财政风险补偿金的8倍发放贷款。在风险补偿上,按照银行实际放贷规模核定财政风险补偿比例,放贷规模越大补偿比例越高,起到了很好的激励约束作用。截至2017年6月末,江西省通过“财政惠农信贷通”累计贷款323.29亿元。江苏、河北、浙江等不少省份都对“银行贷款+
- selenium后续!!
paid槮
selenium测试工具
小项目案例:实现批量下载网页中的资源根据15.3.2小节中的返回网页内容可知,用户只有获取了网页中的图片url才可以将图片下载到*在使用selenium库渲染网页后,可直接通过正则表达式过滤出指定的网页图片,从而实现批量下载接下来以此为思路来实现一个小项目案例。项目任务实现批量下载人民邮电出版社官网中与Python相关的图书封面图片。项目实步骤步骤1,获取人民邮电出版社官网中与Python相关的图
- 参加网络学习收获心得
临江253王馨卉
在七月份以及八月初的几次培训中,我分别接触了不同种类的教育媒介以及教学工具,刷新了我的教学观念。在此我想简单的对几次学习做一个分享。第一部分结合梁校长的讲解,首先刷新了我对PPT应用于教学过程中的认知,以前只觉得这就是代替板书的一种工具,简单明了就行,但是现在认识到设计一个PPT要注意到情境创设,化抽象为直观以及它交互练习的特别作用。根据单页PPT设计的要求,我对字体大小,多少以及颜色都进行了调整
- 2021-10-10
如鱼饮水2020
中原焦点团队网络中26期坚持分享第516天(20211010)论文答辩稿背了N篇,第三个就是我上台演讲,心跳加速,深呼吸极力默念:稳就是定海神针。前两位男生都是低头读稿,虽然自己也带着手稿,距离太远根本看不到。硬着头皮背吧,脑子断篇也得上,先阳谋一下:第一次上讲台特别紧张。调整一下语速,结合APP,能背多少是多少吧,丑媳妇总归要见公婆的。先感谢导师的悉心指导和在坐的各位,论文的结构框架和大概内容几
- 网站开发公司
红匣子实力推荐
随着互联网的普及和发展,越来越多的企业和个人意识到拥有一个专业、高效、易用的网站对于品牌形象和业务拓展的重要性。为了满足这一需求,专业的网站开发公司应运而生,致力于为客户提供一站式的网站建设和优化服务。本文将为您详细介绍网站开发公司的主要业务范围、核心竞争力以及如何选择一家合适的网站开发公司。开发-联系电话:13642679953(微信同号)一、网站开发公司的主要业务范围1.网站策划与设计:根据客
- 力扣—水果成篮
文章目录题目解析解题思路代码实现题目解析你正在探访一家农场,农场从左到右种植了一排果树。这些树用一个整数数组fruits表示,其中fruits[i]是第i棵树上的水果种类。你想要尽可能多地收集水果。然而,农场的主人设定了一些严格的规矩,你必须按照要求采摘水果:你只有两个篮子,并且每个篮子只能装单一类型的水果。每个篮子能够装的水果总量没有限制。你可以选择任意一棵树开始采摘,你必须从每棵树(包括开始采
- 激动!《神秘海域》电影版被爆开拍!
Summer雨农
开发多年的著名游戏改编电影《神秘海域》真的要拍了。主演汤姆·霍兰德近日在宣传新片《1/2的魔法》时告诉IGN:《神秘海域》电影将于4个星期后开拍,那就是3月了。他还着重提到了动作戏,及透露电影灵感很多来自自游戏《神秘海域4》。4480青苹果影院免费在线观看最新电影电视剧。被IGN问到该片,荷兰弟表示:“我们4个星期后就开拍,马克·沃尔伯格演的Sully会很棒的,我们在柏林的特技部门已经做出了很棒的
- 有没有简单的日入1000赚钱途径(掌握日入1000赚钱方法)
幸运副业
有没有简单的日入1000赚钱途径(掌握日入1000赚钱方法)每个人都希望能够找到一种简单的日入1000赚钱途径。虽然没有一种途径能够让你一夜暴富,但是通过一些有效的方法和平台,你完全可以在短时间内实现日入1000的目标。在这篇文章中,我们将为你分享一些实用的赚钱方法和推荐多职猫兼职平台。推荐一篇找兼职必看的免费教程:《手机兼职,300-500/天,一单一结,大量要人》在这里可以找到各种手机截图兼职
- 网络编程中的 Protobuf 和 JsonCpp 全面解析
筏.k
c++asio网络编程网络开发语言c++服务器
文章目录前言一、为什么需要序列化?序列化的好处:常见序列化格式包括:二、JsonCpp与Protobuf对比三、JsonCpp简介与示例(客户端通信)JsonCpp使用示例(客户端发送请求):JsonCpp使用示例(服务器解析请求):四、Protobuf简介与示例(服务器通信)定义消息格式(user.proto)编译生成代码:服务器端序列化&发送数据接收端解析数据五、使用建议总结前言在网络编程中,
- 8.10少阳条辨续与温胆汤
吕思萍
一、柴胡芍药枳实甘草汤与宋本四逆散1、柴胡芍药枳实甘草汤1)主证是邪下痛,然后痛得剧烈的时候人会吐2)胸中气降不下来这个人是右胁痛,上来的气不通的话是左胁痛。治左胁痛以柴胡芍药为主,治右胁痛你枳实为主,都有。2、宋本四逆散1)四逆散,正是一个肝胆之气郁结,胡量少,可以疏肝2)阳气郁结,闷在身体中间不能传到四肢,脉弦,里热舌苔黄,心烦3)逍遥散、柴胡疏肝散,几乎所有的舒肝解郁的方都是从四逆散发展出来
- 2023-09-25
与非与你
01下个星期就要给表哥做伴娘了,再下个星期又要给一个好朋友做姐妹,今年不管是家人还是朋友都很多结婚了,自己的那种无措感又来了。看着别人结婚,自己已经没有当初那种想结婚的欲望了,只是觉得大家都长大了,从读书的年纪突然一下子就到了结婚生子的年纪。昨天和朋友四点半起床开车去海边看日出,一下车听到海浪声,那个风真的自由和快乐。我想如果结婚生孩子了,是否还有这样的机会说走就走的时候?是否还能像现在这么洒脱和
- C#语法基础总结(超级全面)(二)
inwith
C#语法基础c#开发语言
文章目录c#语法基本元素关键字操作符(operator)类型转换标识符(Identifier)语句try语句迭代语句(循环语句)索引器文本(字面值)五大数据类型引用类型:值类型:变量、对象与内存装箱和拆箱类类的实例化类的三大成员(属性、方法、事件)属性(property)方法(函数)方法参数值参数引用参数输出参数数组参数具名参数可选参数扩展方法(this参数)方法的重载构造器(constructo
- Python爬虫博客:使用Selenium模拟登录并抓取需要身份验证的网站内容
Python爬虫项目
2025年爬虫实战项目python爬虫selenium信息可视化开发语言百度测试工具
引言在爬虫开发的过程中,我们常常遇到需要身份验证才能访问的网站。例如,很多社交媒体、新闻网站、电商平台等都要求用户登录才能访问一些特定内容。如何模拟登录并抓取这些需要身份验证的网页内容成为了一个非常重要且常见的需求。Selenium,作为一个强大的浏览器自动化工具,不仅可以模拟用户的浏览行为,还能够模拟用户输入用户名和密码、点击登录按钮等操作,突破了普通爬虫工具(如requests)无法处理的Ja
- jsonp 常用util方法
hw1287789687
jsonpjsonp常用方法jsonp callback
jsonp 常用java方法
(1)以jsonp的形式返回:函数名(json字符串)
/***
* 用于jsonp调用
* @param map : 用于构造json数据
* @param callback : 回调的javascript方法名
* @param filters : <code>SimpleBeanPropertyFilter theFilt
- 多线程场景
alafqq
多线程
0
能不能简单描述一下你在java web开发中需要用到多线程编程的场景?0
对多线程有些了解,但是不太清楚具体的应用场景,能简单说一下你遇到的多线程编程的场景吗?
Java多线程
2012年11月23日 15:41 Young9007 Young9007
4
0 0 4
Comment添加评论关注(2)
3个答案 按时间排序 按投票排序
0
0
最典型的如:
1、
- Maven学习——修改Maven的本地仓库路径
Kai_Ge
maven
安装Maven后我们会在用户目录下发现.m2 文件夹。默认情况下,该文件夹下放置了Maven本地仓库.m2/repository。所有的Maven构件(artifact)都被存储到该仓库中,以方便重用。但是windows用户的操作系统都安装在C盘,把Maven仓库放到C盘是很危险的,为此我们需要修改Maven的本地仓库路径。
- placeholder的浏览器兼容
120153216
placeholder
【前言】
自从html5引入placeholder后,问题就来了,
不支持html5的浏览器也先有这样的效果,
各种兼容,之前考虑,今天测试人员逮住不放,
想了个解决办法,看样子还行,记录一下。
【原理】
不使用placeholder,而是模拟placeholder的效果,
大概就是用focus和focusout效果。
【代码】
<scrip
- debian_用iso文件创建本地apt源
2002wmj
Debian
1.将N个debian-506-amd64-DVD-N.iso存放于本地或其他媒介内,本例是放在本机/iso/目录下
2.创建N个挂载点目录
如下:
debian:~#mkdir –r /media/dvd1
debian:~#mkdir –r /media/dvd2
debian:~#mkdir –r /media/dvd3
….
debian:~#mkdir –r /media
- SQLSERVER耗时最长的SQL
357029540
SQL Server
对于DBA来说,经常要知道存储过程的某些信息:
1. 执行了多少次
2. 执行的执行计划如何
3. 执行的平均读写如何
4. 执行平均需要多少时间
列名 &
- com/genuitec/eclipse/j2eedt/core/J2EEProjectUtil
7454103
eclipse
今天eclipse突然报了com/genuitec/eclipse/j2eedt/core/J2EEProjectUtil 错误,并且工程文件打不开了,在网上找了一下资料,然后按照方法操作了一遍,好了,解决方法如下:
错误提示信息:
An error has occurred.See error log for more details.
Reason:
com/genuitec/
- 用正则删除文本中的html标签
adminjun
javahtml正则表达式去掉html标签
使用文本编辑器录入文章存入数据中的文本是HTML标签格式,由于业务需要对HTML标签进行去除只保留纯净的文本内容,于是乎Java实现自动过滤。
如下:
public static String Html2Text(String inputString) {
String htmlStr = inputString; // 含html标签的字符串
String textSt
- 嵌入式系统设计中常用总线和接口
aijuans
linux 基础
嵌入式系统设计中常用总线和接口
任何一个微处理器都要与一定数量的部件和外围设备连接,但如果将各部件和每一种外围设备都分别用一组线路与CPU直接连接,那么连线
- Java函数调用方式——按值传递
ayaoxinchao
java按值传递对象基础数据类型
Java使用按值传递的函数调用方式,这往往使我感到迷惑。因为在基础数据类型和对象的传递上,我就会纠结于到底是按值传递,还是按引用传递。其实经过学习,Java在任何地方,都一直发挥着按值传递的本色。
首先,让我们看一看基础数据类型是如何按值传递的。
public static void main(String[] args) {
int a = 2;
- ios音量线性下降
bewithme
ios音量
直接上代码吧
//second 几秒内下降为0
- (void)reduceVolume:(int)second {
KGVoicePlayer *player = [KGVoicePlayer defaultPlayer];
if (!_flag) {
_tempVolume = player.volume;
- 与其怨它不如爱它
bijian1013
选择理想职业规划
抱怨工作是年轻人的常态,但爱工作才是积极的心态,与其怨它不如爱它。
一般来说,在公司干了一两年后,不少年轻人容易产生怨言,除了具体的埋怨公司“扭门”,埋怨上司无能以外,也有许多人是因为根本不爱自已的那份工作,工作完全成了谋生的手段,跟自已的性格、专业、爱好都相差甚远。
- 一边时间不够用一边浪费时间
bingyingao
工作时间浪费
一方面感觉时间严重不够用,另一方面又在不停的浪费时间。
每一个周末,晚上熬夜看电影到凌晨一点,早上起不来一直睡到10点钟,10点钟起床,吃饭后玩手机到下午一点。
精神还是很差,下午像一直野鬼在城市里晃荡。
为何不尝试晚上10点钟就睡,早上7点就起,时间完全是一样的,把看电影的时间换到早上,精神好,气色好,一天好状态。
控制让自己周末早睡早起,你就成功了一半。
有多少个工作
- 【Scala八】Scala核心二:隐式转换
bit1129
scala
Implicits work like this: if you call a method on a Scala object, and the Scala compiler does not see a definition for that method in the class definition for that object, the compiler will try to con
- sudoku slover in Haskell (2)
bookjovi
haskellsudoku
继续精简haskell版的sudoku程序,稍微改了一下,这次用了8行,同时性能也提高了很多,对每个空格的所有解不是通过尝试算出来的,而是直接得出。
board = [0,3,4,1,7,0,5,0,0,
0,6,0,0,0,8,3,0,1,
7,0,0,3,0,0,0,0,6,
5,0,0,6,4,0,8,0,7,
- Java-Collections Framework学习与总结-HashSet和LinkedHashSet
BrokenDreams
linkedhashset
本篇总结一下两个常用的集合类HashSet和LinkedHashSet。
它们都实现了相同接口java.util.Set。Set表示一种元素无序且不可重复的集合;之前总结过的java.util.List表示一种元素可重复且有序
- 读《研磨设计模式》-代码笔记-备忘录模式-Memento
bylijinnan
java设计模式
声明: 本文只为方便我个人查阅和理解,详细的分析以及源代码请移步 原作者的博客http://chjavach.iteye.com/
import java.util.ArrayList;
import java.util.List;
/*
* 备忘录模式的功能是,在不破坏封装性的前提下,捕获一个对象的内部状态,并在对象之外保存这个状态,为以后的状态恢复作“备忘”
- 《RAW格式照片处理专业技法》笔记
cherishLC
PS
注意,这不是教程!仅记录楼主之前不太了解的
一、色彩(空间)管理
作者建议采用ProRGB(色域最广),但camera raw中设为ProRGB,而PS中则在ProRGB的基础上,将gamma值设为了1.8(更符合人眼)
注意:bridge、camera raw怎么设置显示、输出的颜色都是正确的(会读取文件内的颜色配置文件),但用PS输出jpg文件时,必须先用Edit->conv
- 使用 Git 下载 Spring 源码 编译 for Eclipse
crabdave
eclipse
使用 Git 下载 Spring 源码 编译 for Eclipse
1、安装gradle,下载 http://www.gradle.org/downloads
配置环境变量GRADLE_HOME,配置PATH %GRADLE_HOME%/bin,cmd,gradle -v
2、spring4 用jdk8 下载 https://jdk8.java.
- mysql连接拒绝问题
daizj
mysql登录权限
mysql中在其它机器连接mysql服务器时报错问题汇总
一、[running]
[email protected]:~$mysql -uroot -h 192.168.9.108 -p //带-p参数,在下一步进行密码输入
Enter password: //无字符串输入
ERROR 1045 (28000): Access
- Google Chrome 为何打压 H.264
dsjt
applehtml5chromeGoogle
Google 今天在 Chromium 官方博客宣布由于 H.264 编解码器并非开放标准,Chrome 将在几个月后正式停止对 H.264 视频解码的支持,全面采用开放的 WebM 和 Theora 格式。
Google 在博客上表示,自从 WebM 视频编解码器推出以后,在性能、厂商支持以及独立性方面已经取得了很大的进步,为了与 Chromium 现有支持的編解码器保持一致,Chrome
- yii 获取控制器名 和方法名
dcj3sjt126com
yiiframework
1. 获取控制器名
在控制器中获取控制器名: $name = $this->getId();
在视图中获取控制器名: $name = Yii::app()->controller->id;
2. 获取动作名
在控制器beforeAction()回调函数中获取动作名: $name =
- Android知识总结(二)
come_for_dream
android
明天要考试了,速速总结如下
1、Activity的启动模式
standard:每次调用Activity的时候都创建一个(可以有多个相同的实例,也允许多个相同Activity叠加。)
singleTop:可以有多个实例,但是不允许多个相同Activity叠加。即,如果Ac
- 高洛峰收徒第二期:寻找未来的“技术大牛” ——折腾一年,奖励20万元
gcq511120594
工作项目管理
高洛峰,兄弟连IT教育合伙人、猿代码创始人、PHP培训第一人、《细说PHP》作者、软件开发工程师、《IT峰播》主创人、PHP讲师的鼻祖!
首期现在的进程刚刚过半,徒弟们真的很棒,人品都没的说,团结互助,学习刻苦,工作认真积极,灵活上进。我几乎会把他们全部留下来,现在已有一多半安排了实际的工作,并取得了很好的成绩。等他们出徒之日,凭他们的能力一定能够拿到高薪,而且我还承诺过一个徒弟,当他拿到大学毕
- linux expect
heipark
expect
1. 创建、编辑文件go.sh
#!/usr/bin/expect
spawn sudo su admin
expect "*password*" { send "13456\r\n" }
interact
2. 设置权限
chmod u+x go.sh 3.
- Spring4.1新特性——静态资源处理增强
jinnianshilongnian
spring 4.1
目录
Spring4.1新特性——综述
Spring4.1新特性——Spring核心部分及其他
Spring4.1新特性——Spring缓存框架增强
Spring4.1新特性——异步调用和事件机制的异常处理
Spring4.1新特性——数据库集成测试脚本初始化
Spring4.1新特性——Spring MVC增强
Spring4.1新特性——页面自动化测试框架Spring MVC T
- idea ubuntuxia 乱码
liyonghui160com
1.首先需要在windows字体目录下或者其它地方找到simsun.ttf 这个 字体文件。
2.在ubuntu 下可以执行下面操作安装该字体:
sudo mkdir /usr/share/fonts/truetype/simsun
sudo cp simsun.ttf /usr/share/fonts/truetype/simsun
fc-cache -f -v
- 改良程序的11技巧
pda158
技巧
有很多理由都能说明为什么我们应该写出清晰、可读性好的程序。最重要的一点,程序你只写一次,但以后会无数次的阅读。当你第二天回头来看你的代码 时,你就要开始阅读它了。当你把代码拿给其他人看时,他必须阅读你的代码。因此,在编写时多花一点时间,你会在阅读它时节省大量的时间。
让我们看一些基本的编程技巧:
尽量保持方法简短
永远永远不要把同一个变量用于多个不同的
- 300个涵盖IT各方面的免费资源(下)——工作与学习篇
shoothao
创业免费资源学习课程远程工作
工作与生产效率:
A. 背景声音
Noisli:背景噪音与颜色生成器。
Noizio:环境声均衡器。
Defonic:世界上任何的声响都可混合成美丽的旋律。
Designers.mx:设计者为设计者所准备的播放列表。
Coffitivity:这里的声音就像咖啡馆里放的一样。
B. 避免注意力分散
Self Co
- 深入浅出RPC
uule
rpc
深入浅出RPC-浅出篇
深入浅出RPC-深入篇
RPC
Remote Procedure Call Protocol
远程过程调用协议
它是一种通过网络从远程计算机程序上请求服务,而不需要了解底层网络技术的协议。RPC协议假定某些传输协议的存在,如TCP或UDP,为通信程序之间携带信息数据。在OSI网络通信模型中,RPC跨越了传输层和应用层。RPC使得开发