map与alignment
新一代测序数据的处理过程中,map/alignment总是同时出现,那么二者的区别是什么呢?大致上可以这样理解:map这个动作表示的是:把短片段对应到参考基因组的某个位置上。这个动作只回答“短片段来自长片段的什么位置?”这个问题;而align这个动作早在二代测序之前很久就有了,这个动作表示两条(或多条)序列(不管是核酸序列还是蛋白序列)的比较,回答的问题是“这两条序列之间有什么异同?”。显然在处理重测序数据的过程中,要先map,再align,即先把read定位到参考基因组的某个位置上,再比较read和参考基因组序列的异同。另外还可以认为:凡是要align,那一定是要先map的;但是map过后,却不一定要align(比如我只关注CNV或表达量等信息,而不关心具体序列的时候,就只需要map确定下read来自参考序列的哪个位置,而不需要align获取具体的序列间异同信息)。可见在SAM文件中,map和align这两个动作都要有,这可能就是这两个动作总是同时出现的原因。理解了这两个名字的含义和区别,就可以理解为什么是“mapping” position,因为确定position信息只需要mapping。map和align两个词,似乎都可以用中文“比对”一词来表示。下文中如果没有必要区分二者,将全都译作“比对”。
bwa
命令算法:
bwa
bwa index 建立索引
bwa mem BWA_MEM algorithm
bwa bwasw BWA_SW for long queries
bwa aln gapped/ungapped alignment
bwa samse generate alignment(single ended)
bwa sampe generate alignment(paired ended)
可通过bwa index/bwa mem/bwa aln查看相应命令的帮助
比对流程
-建立一个已知参考基因组的索引
索引的建立是对参考基因组进行重新格式化,每个比对软件的索引不同。基因组越大,构建索引的时间越长。很多比对工具在其官方网站上有提供已经预建的索引文件,可直接下载使用。
-FASTA/FASTQ文件中的reads比对到索引上
示例
参照原文用ebola病毒测序数据和1976年Mayinga参考基因组。
mkdir -p ~/refs/ebola #建立文件夹用于存放参考基因组文件及将建立的索引存放于此
下载ebola病毒参考基因组文件,命名为1976.fa
efetch -db=nuccore -format=fasta -id=AF086833 > ~/refs/ebola/1976.fa
建立索引
bwa index ~/refs/ebola/1976.fa
还好前段时间看了下shell编程,才看懂文中的变量的用法,基础差的学生就得想办法恶补。
REF=~/refs/ebola/1976.fa #将后面的路径赋值给变量REF
bwa index $REF #shell中用$加变量名用来引用变量值
ls ~/refs/ebola #查看建立索引后目录内文件的变化
1976.fa 1976.fa.amb 1976.fa.ann 1976.fa.bwt 1976.fa.pac 1976.fa.sa
获取SRR1972739的ebola病毒的测序数据
esearch -db sra -query PRJNA257197 | efetch -format runinfo > runinfo.csv
fastq-dump -X 10000 --split-files SRR2571197#通过fastq-dump命令将sra数据还原为fastq文件,双端测序文件加上参数--split-files
命令结束后会在当前目录下产生两个测序数据
SRR1972739_1.fastq和SRR1972739_2.fastq
R1=SRR1972739_1.fastq #定义定量R1
R2=SRR1972739_2.fastq #定义变量R2
执行比对
bwa mem $REF $R1 $R2 > bwa.sam
bwa mem
bowtie
Bowtie 2 is an ultrafast and memory-efficient tool for aligning sequencing reads to long reference sequences. It is particularly good at aligning reads of about 50 up to 100s or 1,000s of characters, and particularly good at aligning to relatively long (e.g. mammalian) genomes. Bowtie 2 indexes the genome with an FM Index to keep its memory footprint small: for the human genome, its memory footprint is typically around 3.2 GB. Bowtie 2 supports gapped, local, and paired-end alignment modes.
算法可参考天天向上的博客
工作流程
-建立索引
-将FASTA/FASTQ格式的reads与索引进行比对
bowtie2 -h
Usage:
bowtie2 [options]* -x{-1 -2 | -U } [-S ]
Index filename prefix (minus trailing .X.bt2).
NOTE: Bowtie 1 and Bowtie 2 indexes are not compatible.
Files with #1 mates, paired with files in .
Could be gzip'ed (extension: .gz) or bzip2'ed (extension: .bz2).
Files with #2 mates, paired with files in .
Could be gzip'ed (extension: .gz) or bzip2'ed (extension: .bz2).
Files with unpaired reads.
Could be gzip'ed (extension: .gz) or bzip2'ed (extension: .bz2).
File for SAM output (default: stdout)
, , can be comma-separated lists (no whitespace) and can be
specified many times. E.g. '-U file1.fq,file2.fq -U file3.fq'.
建立索引文件
bowtie2-build -h
Usage: bowtie2-build [options]*
reference_in comma-separated list of files with ref sequences
bt2_index_base write bt2 data to files with this dir/basename
*** Bowtie 2 indexes work only with v2 (not v1). Likewise for v1 indexes. ***
Options:
-f reference files are Fasta (default)
-c reference sequences given on cmd line (as
)
--large-index force generated index to be 'large', even if ref
has fewer than 4 billion nucleotides
-a/--noauto disable automatic -p/--bmax/--dcv memory-fitting
-p/--packed use packed strings internally; slower, less memory
--bmaxmax bucket sz for blockwise suffix-array builder
--bmaxdivnmax bucket sz as divisor of ref len (default: 4)
--dcvdiff-cover period for blockwise (default: 1024)
--nodc disable diff-cover (algorithm becomes quadratic)
-r/--noref don't build .3/.4 index files
-3/--justref just build .3/.4 index files
-o/--offrateSA is sampled every 2^ BWT chars (default: 5)
-t/--ftabchars# of chars consumed in initial lookup (default: 10)
--threads# of threads
--seedseed for random number generator
-q/--quiet verbose output (for debugging)
-h/--help print detailed description of tool and its options
--usage print this usage message
--version print version information and quit
bowtie2-build $REF $REF #建立索引
第一个路径表示参考文件,第二个表示索引的前缀名称,可以是anything。
ls $REF.*
运行比对
bowtie2 -x $REF -1 $R1 -2 $R2 > bowtie.sam
比对软件的比较与选择
SAM文件

SAM代表Sequence Alignment/Map格式,是一种制表符分隔的文本格式,包含一个可选的头部分(header section,有人称之为“注释部分”),和一个比对部分(alignment section)。如果包含头部分,那么头部分必须置于比对部分之前。头部分的行以@符号开头,而比对部分的行不以@符号开头。比对部分的每一行包含11个必选的字段,用于说明重要的比对信息,如比对位置(mapping position)等;另有可变数量的可选字段,用于存储其他信息(flexible)或比对软件特异的信息。
header部分
@开头,后面紧接着一个或两个字母的头记录类型编码。每行都是以制表符分隔的,除了@CO行,每个数据字段都服从‘TAG:VALUE’这一格式,其中TAG是一个两字符的字符串,定义了VALUE的格式和内容。所以头部分的行都符合这一正则表达式: /^@[A-Z][A-Z](\t[A-Za-z][A-Za-z0-9]:[ -~]+)+$/或者 /^@CO\t.*/
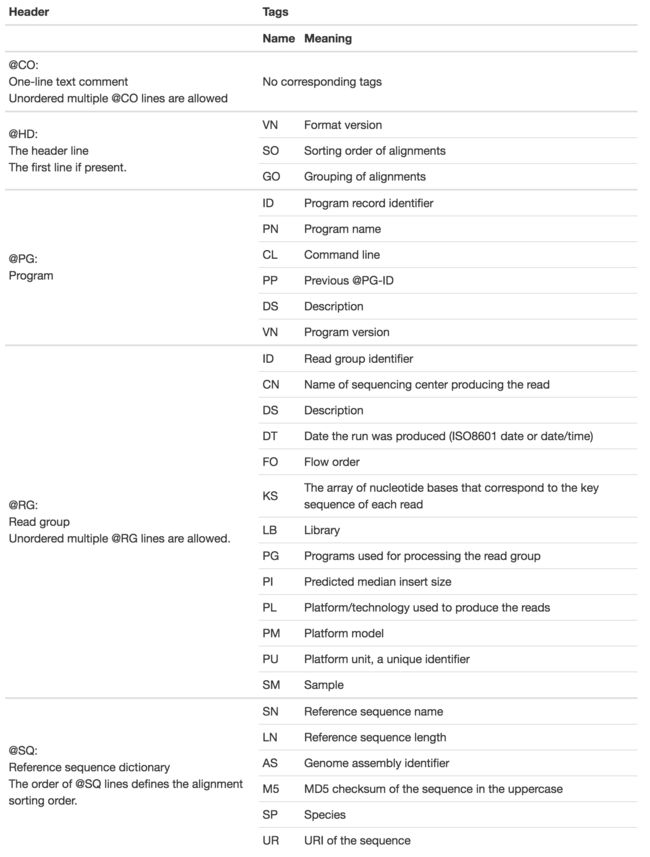
比对部分
每行表示一个片段(segment)的线性比对(linear alignment)。每行有11个必选字段,信息缺失字段用0或者“*”表示。
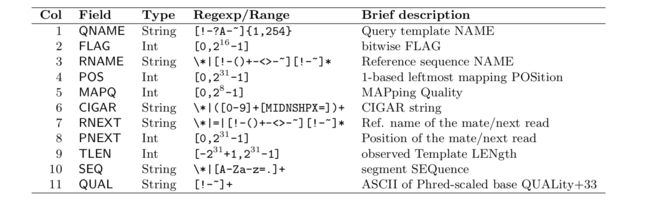
FLAG字段的理解
SAM 文件是比对结果的展示, 格式详细介绍见官方文档, 其中第二列为按位标记的 flag, 这是一种常用且高效的保存多个布尔特征值的方法, 可通过按位取 '与' 和取 '或' 快速提取对应的值
举个简单的例子: 在 SAM 格式中, 当 flag 为 1, 也即对应的二进制为 01 时, 表示该 read 有多个测序数据 (template having multiple segments in sequencing), 一般理解为有双端测序数据 (另一条没被过滤掉), 而 flag 为 2, 也即二进制 10 时, 表示这条 read 的多个片断都有比对结果 (each segment properly aligned according to the aligner), 通常理解为双端 reads 都比对上了, 那么就可以推断出 flag 为 3 时, 也即二进制的 11, 表示该 read 有另一端的 read 并且比对成功, 可以看到, 其实就是 01 加 10, 反之, 当我们看到二进制的 11, 就可以想到这表示这两位的状态均为 1 (通常即为真), 再查询定义就知道了具体的含义
这样就通过一个位来反映一个 True/ False 的状态值, 同时方便了人脑和电脑, 在 SAM flag 中, 目前共有 12 种不同状态, 列举如下:
(来源:http://not.farbox.com/post/sam_flag)
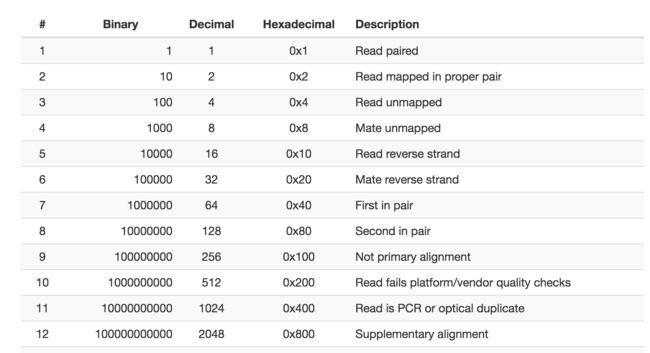
SAM Format提供了工具可快速查询各个value的含义。
将其按需求自由组合, 再结合使用 samtools 软件 view 命令的 -f (满足) -F (不满足) 和 -c (计数) 参数, 就可以实现各种过滤和统计了, 比如 samtools view -f 4 就会提取出未比对上的 reads, samtools view -c -F 4 就能统计出比对上的 reads 数, 也即比对率的分子部分
CIGAR字段
