本文内容来自文献: Cancer Genome Landscapes

目前已发现近140种“driver”基因
driver gene:给予一种选择性生长优势,促进或驱动肿瘤发生。一种这样的生长优势造成的差异很小,但会累积,最终造成很大的混乱,包含数十亿细胞。
driver gene可被分进3种核心细胞过程, cell fate(细胞命运)、 cell survival(细胞生存)、 genome maintenance(基因组维稳)中的12种信号途径中。
一个常见肿瘤含2~8种“driver” gene突变,其它的gene突变称为“passenger” gene突变。
passenger gene:没有选择性生长优势。
一、How many genes are subtly mutated in a typical human cancer?
①一般实体瘤:平均33~36种gene突变。95%为单碱基突变,剩下的是缺失或插入一个或几个碱基。在碱基替换中,90.7%为错义突变,7.6%为无义突变,1.7%导致起始密码子和终止密码子相临的剪接位点或未翻译区域的改变。
②突变种类比平均水平高很多:如肺癌和黑色素瘤含约200种非无义突变,这种大数量突变可能有两种可能。a、有致突变剂的参与(如紫外线和抽烟等)b、DNA修复功能障碍。现在的研究表明有大量的突变发生在聚合酶POLE或POLD1的矫正域。
POLE参考
POLD1参考
③突变种类比平均水平低很多:如小儿肿瘤和血瘤的点突变数量则会少很多,平均9.6个突变每个肿瘤。
二、Mutation Timing
以大肠直肠癌中,肿瘤从良性变成恶性的过程举例:
a、守门突变(getakeeping mutation),常发生在APC基因。给一个正常上皮细胞提供选择性生长优势,在它周围形成微小克隆。
APC基因参考
b、KRAS基因发生突变。导致克隆数增多,仅APC突变的数目明显少于APC和KRAS都突变的。
KRAS基因参考
c、随着克隆扩增,一系列基因突变,如PIK3CA、SMAD4和P53基因突变,最终形成恶性肿瘤扩散至淋巴结或远处的组织。
PIK3CA参考
PIK3CA参考
PIK3CA参考
SMAD4参考
P53参考

自我更新(复制分裂)组织中某些肿瘤的突变数量与年龄直接相关,线性回归分析表明这些肿瘤中超过一半的体细胞突变发生在肿瘤发生前阶段,这些癌前突变都是对肿瘤过程没有影响的“passenger”突变。这解释了:①同种自我更新组织的肿瘤(比如直肠癌 colorectal tumor 、),90岁的老人比45的患者有将近两倍的突变。②自我更新组织的肿瘤(如脑肿瘤中的神经胶母细胞瘤glioblastomas、胰腺导管腺癌pancreatic ductal adenocarcinomas)比自我更新组织的肿瘤含有的突变要少,相应的它们的肿瘤发生的前体细胞中含有的突变也自然更少。③儿童癌症的突变比成人肿瘤少。儿童癌症通常发生在非自我更新的组织中,而那些出现在自我更新的组织(如白血病leukemias)中的癌症的前体细胞没有像成人那样经常自我更新。此外,与成人实体瘤相比,儿童肿瘤以及成人白血病和淋巴瘤 lymphomas可能需要较少轮的克隆扩张。
肿瘤中体细胞突变的数量的钟clock表明:首先,需要几十年的时间发展成一种成熟的转移性癌症。其次,原发性肿瘤中已经有大量细胞出现转移病灶的实质转移瘤。
从基因的视角看,早期肿瘤到转移性肿瘤之间肯定又发生了突变,就像正常细胞变成良性肿瘤,或者良性肿瘤变成恶性肿瘤那样。一种可能的解释是,突变或表观遗传变化很难用目前的技术识别(见下文“暗物质”)。另一种解释是,转移性病变尚未得到足够详细的研究,以确定这些基因改变,特别是在突变性质是异质的情况下。但另一种可能的解释是没有转移基因。恶性原发性肿瘤的转移可能需要数年时间,但这一过程原则上仅由随机过程来解释。晚期肿瘤每天向循环系统释放数百万个细胞,但这些细胞的半衰期很短,只有一小部分细胞发生转移。可以想象,这些循环细胞可能以一种不确定的方式,不经常地、随机地滞留在器官的毛细血管床中,为生长提供了有利的微环境。原发肿瘤肿块越大,发生这一过程的可能性越大。在这种情况下,原发性肿瘤的持续发展将反映局部选择优势,而不是未来的选择优势。最近的研究结果表明,即使是正常的细胞,在适当的环境下,如淋巴结,也能长成具有正常血管功能的类器官。
三、Other Types of Genetic Alterations in Tumors
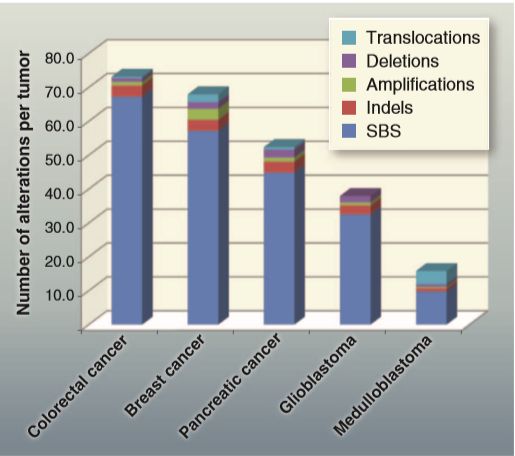
虽然肿瘤的点突变率与正常细胞的点突变率相似,但肿瘤的染色体变化率却升高了。因此,大多数实体瘤在染色体数目(非整倍性)、缺失、倒位、易位以及其他基因异常等方面表现出广泛的改变。当染色体的一大部分被复制或删除时,很难确定染色体上的得失对肿瘤细胞具有生长优势的特定“靶”基因。在染色体易位、纯合子缺失和基因扩增的情况下,目标基因更容易识别。易位通常融合两个基因来产生一个致癌基因(如慢性粒细胞白血病中的BCR-ABL),但在少数情况下,可以通过截断或从启动子中分离抑癌基因而使其失活。纯合子缺失通常只涉及一个或几个基因,其靶点始终是抑癌基因。扩增包含一种原癌基因,其蛋白产物异常活跃,肿瘤细胞中每个细胞含有10到100个该基因的拷贝,而正常细胞中只有两个拷贝。
非整倍性参考
BCR-ABL参考
纯合子缺失
大多数实体瘤有几十个易位;然而,与点突变一样,大多数易位似乎是“passenger”而不是“driver”。易位的断点通常在缺乏已知基因的“gene deserts”中,许多易位和纯合子缺失与易破裂的脆弱部位相邻。这样的染色体断裂发生时,癌细胞可能比正常细胞更容易存活,因为它们含有突变,比如使像TP53这样的基因丧失能力(DNA损伤时,TP53通常会通过引发细胞死亡)。迄今为止的研究表明,受染色体变化影响的基因大约是受点突变影响的基因的10倍。下图显示了影响蛋白质编码基因的5种典型肿瘤类型的遗传改变的类型和分布。蛋白质编码基因仅占总基因组的约1.5%,非编码区的变化数比影响编码区的变化数成比例地高。非编码区的绝大多数变化可能是“passenger”,这些非编码突变以及在癌症中发现的大量表观遗传变化将在后面讨论。
四、Drivers Versus Passenger Mutations
很难确定哪些体细胞突变是“driver”,哪些是“passenger”。此外,“driver”基因和“driver”基因突变之间存在着根本区别。“driver”基因是包含“driver”基因突变的基因。但“driver”基因也可能包含“passenger”基因突变。例如,APC是一个大的“driver”基因,但只有那些截断了其N末端1600个氨基酸以内的编码蛋白的突变才是“driver”基因突变。整个基因的错义突变,以及C末端1200个氨基酸的蛋白截断突变,都是“passenger”基因突变。
有许多统计方法来确定“driver”基因。一些是基于单个基因的突变频率,在校正了序列背景(周边序列)和基因大小后,与同一肿瘤或相关肿瘤中其他基因的突变频率相比。其他方法是基于预测突变对编码蛋白的影响,根据生物物理学进行推断。所有这些方法都有助于对最有可能的"driver"基因进行优先排序。当一个基因的突变数量非常高时,如TP53或KRAS,任何合理的统计都将表明该基因极有可能是一个驱动基因。这些高度突变的基因被称为“mountains”。然而,在癌症基因组中占主导地位是具有一个以上但相对较少突变数量的基因(称为“hills”)。在这种情况下,仅仅基于突变频率和背景的方法不能可靠地指出哪些基因是“driver”,因为基因突变的背景率在不同的病人和基因组的不同的区域变化很大。最近对正常细胞的研究表明,突变率在基因组中的变化超过100倍。在肿瘤细胞中,这种变异可能更高,并且可能以一种明显随机的方式影响整个基因组区域。因此,基于突变频率的方法最多只能优先考虑基因进行进一步分析,但不能明确地识别在相对较低频率突变的“driver”基因。
癌症文献中使用的术语“driver gene”有两个截然不同的含义。“driver”与“passenger”的概念最初被用来区分导致选择性生长优势的突变与未导致选择性生长优势的突变。根据这个定义,不携带“driver”基因突变的基因不能是“driver”基因。但是许多包含很少或没有驱动基因突变的基因在文献中被标记为驱动基因。这些基因包括在肿瘤中过度表达、表达不足或表观遗传学改变的基因,或在实验操纵其表达时增强或抑制肿瘤发生的某些方面的基因。尽管这些基因的一部分可能确实在肿瘤过程中起着重要作用,但将它们都笼统地作为“driver”基因有待商榷。为了调和“driver”基因的两个内涵,建议将可能增加肿瘤细胞选择性生长优势的基因分为“Mut-driver”基因或“Epi-driver”基因。

五、A Ratiometric Method to Identify and Classify Mut-Driver Genes
识别“Mut-driver”基因的最好方法是通过突变模式,而不是通过突变频率。原癌基因和抑癌基因的突变模式具有高度的特征性和非随机性。原癌基因在同一个氨基酸位置重复突变,而抑癌基因则通过截短蛋白质的形式突变。
根据这些突变模式而非频率,我们可以确定在COMSIC(Catalogue of Somatic Mutations in Cancer)数据库中记录的总计404863个微小突变的18306个突变基因中,哪些是“Mut-driver”基因,以及它们是否可能作为原癌基因或抑癌基因发挥作用。原癌基因:只需要记录的基因突变中超过20%处于复发位置,并且是错义的。抑癌基因:类似地要求基因中超过20%的记录突变是失活的(inactivating)。这个“20/20规则”是宽泛的,因为所有有良好记录的癌症基因远远超过了这些标准。
一下列举一些20/20规则的应用。当在脑肿瘤中首次发现IDH1突变时,其在肿瘤发生中的作用是未知的。最初的功能研究表明IDH1是一个抑癌基因,突变使该基因失活。然而,几乎所有的IDH1突变都在相同的氨基酸,密码子132。根据20/20规则评估,这种分布明确表明IDH1是一个原癌基因而不是抑癌基因,这一结论最终得到了生化实验的支持。另一个例子是NOTCH1突变。在这种情况下,一些功能性研究表明NOTCH1是一种原癌基因,而另一些研究表明NOTCH1是一种抑癌基因。这种情况可以通过将20/20规则确定,在淋巴瘤和白血病等“液体肿瘤”中,NOTCH1突变经常复发,并且没有截断蛋白质。而在鳞状细胞癌(squamous cell carcinomas)中,NOTCH1突变不是复发性的,通常是失活的。因此,基因数据清楚地表明NOTCH1在不同的肿瘤类型中的功能不同。同一基因在不同细胞类型中以完全相反的方式发挥作用的观点对于理解细胞信号通路很重要。
IDH1参考
六、How Many Mut-Driver Genes Exist?
根据20/20规则定义的,迄今为止仅发现了125个“Mut-driver”基因。其中,71个是抑癌基因,54个是原癌基因。相同的“driver”基因在不同的肿瘤类型中不断被“重新发现”。例如,MLL2和MLL3突变最初在髓母细胞瘤中发现,随后在非霍奇金淋巴瘤、前列腺癌、乳腺癌和其他肿瘤类型中发现。同样的,ARID1A突变首先在透明细胞卵巢癌中被发现,随后在包括胃和肝脏在内的其他几个器官的肿瘤中被发现突变。在最近对几种类型肺癌的研究中,几乎所有被发现有显著突变频率的基因都已在其他器官的肿瘤中被鉴定出来。即经常改变的“Mut-driver”基因("mountains")的数量接近饱和。毫无疑问,会发现更多的“mountains”,但这些“mountains”很可能存在于尚未深入研究的罕见肿瘤类型中。
通过全基因组测序检测到的最新发现的“Mut-driver”基因通常具有启发性。例如,近一半的这些基因编码的蛋白质通过修饰组蛋白或DNA来直接调节染色质。如组蛋白HIST1H3B和H3F3A,以及蛋白质DNMT1和TET1,其共价修饰DNA,EZH2,SETD2和KDM6A。或反过来,甲基化或去甲基化组蛋白。编码mRNA剪接因子的基因,如SF3B1和U2AF1的基因发生了同样的变化,这些基因的突变将导致过多的非特异性细胞应激,而不是促进特定的肿瘤类型。另一个例子是协同蛋白ATRX和DAXX的突变。这些基因突变的肿瘤都有一种特殊类型的端粒延伸过程,称为“ALT”(对于“端粒的选择性延伸”)。最后一个例子是IDH1和IDH2,它们的突变刺激了肿瘤代谢的新兴领域,并对表观遗传学产生了重要影响。
剪接因子参考
“Mut-driver”基因受碱基替换、基因内插入或缺失等细微突变的影响。“Mut-driver”基因也可以通过更大的变化来改变,如易位、扩增和大规模缺失。与点突变一样,很难根据这些类型的突变区分“Mut-driver”基因与仅包含“passenger”突变的基因。目前增加了13个“Mut-driver”基因,10个原癌基因(进行扩增的)和3个抑癌基因(进行纯合删除)到前面所述的125个“Mut-driver”基因,即目前共发现138个“Mut-driver”基因。这13个基因属于非点突变但反复扩增(如MYC家族基因)或者纯合删除(如MAP2K4)并且符合其他标准(如扩增子或纯合删除区域中唯一的基因)。
扩增子参考
很大程度上存在在生殖系中以突变形式遗传而非通过体细胞突变的易致癌的基因,当这些基因异常时,它们通常不会增加选择性生长优势,但它们会直接刺激肿瘤的发生(如通过增加遗传不稳定性)。
七、Dark Matter
在儿童肿瘤如髓母细胞瘤中,"driver"基因突变的数量较低(0到2个)。在常见的成人肿瘤,如胰腺癌、结直肠癌、乳腺癌和脑癌中,突变的驱动基因的数量通常为3到6个,但一些肿瘤只有一个或两个"driver"基因突变。鉴于广泛接受的肿瘤发展和进展需要几十年来获得多个的相继的基因改变的观点,如何解释这一点?
首先是技术性问题。①全基因组测序还远远不够完美,至少在当今的技术条件下。基因组的某些区域没有很好地展现出来。②在序列数据中观察到特定基因的特定核苷酸的次数通常分布广泛,因此某些区域仅用偶然因素无法很好地表示。③原发性肿瘤不仅包含肿瘤细胞,还包含基质细胞,这些基质细胞稀释了突变碱基的信号,进一步降低了发现突变的可能性。
基质细胞参考
技术性问题使得通过NGS手段预测的“driver”基因存在假阴性,并且与外显子测序相比,全基因组测序的假阴性更高,因为后者的序列覆盖率通常低于前者(全基因组研究中通常为30倍,外显子研究中超过100倍)。
观念性问题也限制了可检测“driver”基因的数量。几乎所有的研究,无论是全基因组还是全外显子水平,都聚焦于编码区。当然当驱动基因突变定性地改变编码区的序列时,已经很难识别它们了,而基因间或内含子突变则更难研究。因此,这使一些突变成为无法识别的“暗物质”,甚至在遗传病例的生殖系基因组中。第一个关于非编码的例子:编码端粒酶催化亚单位的TERT基因的启动子的反复突变已经被鉴定出来,并被证明能够激活其转录。
暗物质 ncRNA参考
随着全基因组测序的继续,更多“Mut-driver”基因无疑会被发现。根据上述趋势,大多数“Mut-driver”基因可能是罕见肿瘤类型中的“mountains”或常见肿瘤类型中的“hills”。因此,这些基因不太可能解释大部分假定的暗物质。拷贝数的改变在癌症中普遍存在,无论是在整个染色体还是在部分染色体水平,这些改变都能微妙地改变其“driver”基因的表达。此前的研究表明,丢失一个含有几个抑癌基因的染色体拷贝,每个基因都可能与肿瘤有关,但不因突变而改变,这可能提供选择性生长优势。
拷贝数变异 CNV
暗物质最明显的来源是“Epi-driver“基因。人类肿瘤含有大量影响DNA或染色质蛋白质的表观遗传变化。之前对结直肠癌的一项研究表明,超过10%的蛋白质编码基因与正常的结直肠癌上皮细胞相比,甲基化存在差异。其中一些变化(即”Epi-driver“基因中的变化)可能提供选择性生长优势。例如,CDK2NA和MLH1的表观遗传沉默比这两个公认的"driver"基因中的任何一个的突变失活更为常见,然而,基因的变化和表观遗传变化之间存在一个关键性差异。与特定个体中的基因序列不同,甲基化是可塑的,随细胞类型、发育阶段和患者年龄而变化。启动肿瘤发生的正常前体细胞的甲基化状态尚不清楚。这种可塑性也意味着甲基化可以在微环境信号下发生变化,例如那些与低营养浓度或异常细胞接触有关的信号。因此,很难知道在癌细胞中观察到的特定表观遗传变化是反映还是促成肿瘤状态。区分具有选择性生长优势的表观遗传变化和不具有选择性生长优势的表观遗传变化(”passenger“表观遗传变化)的标准尚未制定。
MLH1参考
八、Genetic Heterogeneity
亚克隆(即肿瘤内的异质性)突变对于理解肿瘤的进化很重要。四种类型的遗传异质性与肿瘤发生有关:
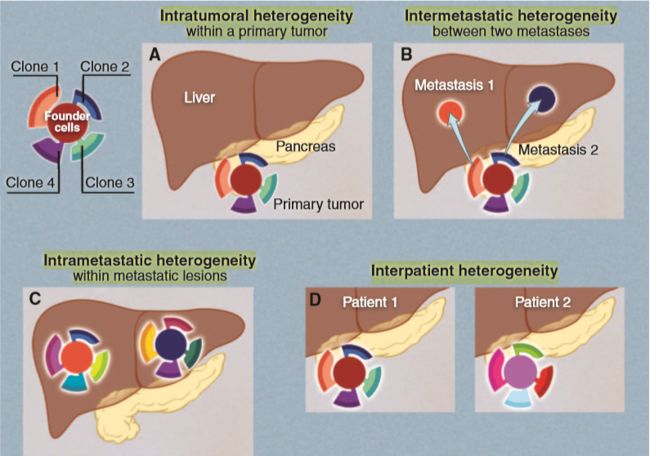
①肿瘤内(intratumoral):一个肿瘤细胞间的异质性。很少看到一个实体肿瘤的细胞遗传学研究,其中所有的肿瘤细胞显示相同的核型。同样的现象也出现在单个基因上,在整个基因组中都有观察到。这种异质性必须存在:每当一个正常(或肿瘤)细胞分裂时,它会获得一些突变,而区分任何两个细胞的突变数量仅仅标志着与上一个共同祖先(其创始细胞)的时间。大肿瘤两端的细胞在空间上是不同的,一般来说,与邻近的细胞相比,两端的细胞间的差异更大。这种现象类似于物种形成,不同岛屿上的有机体比同一岛屿上的有机体更有可能彼此分化。
在通过全基因组测序评估肿瘤内异质性的研究中,大多数体细胞突变出现在所有肿瘤细胞中。这些突变形成了体细胞进化树的主干。分支突变的重要性是什么(即那些不被所有肿瘤细胞共享的突变)?从医学角度来看,这些突变通常毫无意义,因为原发肿瘤是可通过手术切除的。手术前各分支有多少异质性并不重要。然而,这种异质性为转移性间(intermetastastic)异质性提供了种子,具有重要的临床意义。
核型参考
②转移肿瘤间(intermetastastic)异质性(肿瘤间异质性):同一患者不同转移灶的异质性。绝大多数癌症患者死亡是因为他们的肿瘤在转移到手术无法到达的部位,如肝、脑、肺或骨之前没有被切除。复发为单一转移性病变的患者通常仍可以通过手术或放疗治愈,但单一转移是例外,而不是常规。一个临床试验中的典型病人有十几个或更多的转移性病灶,这些病灶大到可以通过影像学观察到,而且更多的转移性病灶更小。如果一个病人的每一个转移性病灶都是由一个基因结构非常不同的细胞所建立的,那么化疗治疗几乎是不可能实现的:根除一个病人的转移性病灶的一个子集不足以长期存活。
不同的转移性病灶有多少异质性?很多。在同一患者中,一个转移性病灶有20个克隆基因改变不被其他转移所共有的情况并不少见。因为它们是克隆的,这些突变发生在转移的创始细胞中,也就是说,从原发肿瘤中逃逸并增殖形成转移的细胞。如预期的那样,每个转移的创始细胞存在于原发肿瘤不同的、地理上不同的区域。
这种潜在的灾难性情况因异质性似乎主要局限于“passenger”基因突变而有所缓和。在大多数记录恶性肿瘤异质性的研究中,“Mut-driver”基因存在于树干中,尽管有例外。这些发现与上述观点一致,即转移所需的基因改变在转移实际发生之前就已经存在。这些数据也与观察结果一致,即在对靶向药物有反应的患者中,反应常常出现在所有转移性病灶中,而不仅仅是一小部分。
③转移肿瘤内(intrametastastic):单个转移肿瘤内的异质性。每一种转移都是由一个单细胞(或一小群细胞)和一系列初始突变形成的。随着肿瘤的生长,每一个细胞分裂都会产生新的突变。尽管创始突变可能使病灶易受抗肿瘤药物的影响,但新的突变为耐药性提供了种子。与原发性肿瘤不同,转移性病灶一般不能通过手术切除,必须进行全身治疗。对靶向治疗有完全反应的患者总是复发。大多数最初的病灶通常复发,并且复发的时间范围明显相似。这一时间过程可以解释为在靶向治疗开始前,每个转移中都存在耐药突变。计算表明,任何在医学成像上可见大小的转移性病灶都有数千个细胞(在数十亿个细胞中),这些细胞几乎对任何可以想象的药物都有抵抗力。因此,复发只是一个时间问题,完全可以根据已知的突变频率和肿瘤细胞生长率预测。原则上,这种“既成事实”可以通过多种药物治疗来规避,因为单个肿瘤细胞不太可能对作用于不同靶点的多种药物产生耐药性。
④病人间异质性(interpatient):不同患者肿瘤的异质性。没有两位癌症患者在接受或不接受治疗的情况下有相同的临床疗程。其中一些差异可能与宿主因素有关,例如决定药物半衰期或血管对药物或细胞的通透性的种系变异,而一些差异可能与非遗传因素有关。然而,这种患者间的异质性可能与肿瘤内的体细胞突变有关。尽管两名患者的乳腺癌中可能存在几十个体细胞突变,但只有少部分是发生在相同的基因,且在绝大多数情况下,这些是“Mur-driver”基因。即使在这些“driver”基因中,实际的突变也往往是不同的。如实验证实的那样,蛋白质不同区域的突变对细胞性质的影响肯定不会相同。尽管相邻密码子的不同突变可能有相同的效果,但对大量患者的详细研究表明,情况并非如此。例如,KRAS的Gly12→Asp12(G12D)突变与同一基因的G13D突变没有相同的临床意义。患者间的异质性一直是设计统一有效治疗癌症的主要障碍之一。基于癌症患者基因组知识的个体化治疗的努力主要基于对这种异质性的认识。
九、Signaling Pathways in Tumors
即使是晚期肿瘤也并非完全失控,这一点可以从针对黑色素瘤突变BRAF或肺癌突变ALK的药物的剧烈反应中得到证明。尽管是短暂的,但这些反应意味着即使是单个突变基因产物的干扰也足以阻止癌症的发展,至少是短暂的。癌症的基因组复杂性如何与这些临床观察相结合呢?
BRAF参考
BRAF参考
ALK参考
这一点有两个概念。首先,上述提到的是,超过99.9%的肿瘤改变(包括点突变、拷贝数改变、易位和表观遗传改变,这些是分布在整个基因组中的,而不仅仅是编码区域)对肿瘤不重要。它们只是标志着连续克隆扩张之间所经过时间的“passenger”变化。正常细胞在分裂时也会发生基因改变,包括核苷酸和染色体水平。然而,正常细胞被编程为细胞死亡来应对这种变化。相比之下,癌细胞通过获得诸如TP53等基因的突变而进化到能够耐受基因组复杂性。因此,基因组的复杂性在一定程度上是癌症的结果,而不是病因。
138个“driver”基因中的所有突变都有个结果:直接或间接地引起选择性生长优势。此外,似乎只有有限数量的细胞信号通路可以产生生长优势。所有已知的驱动基因可分为12个途径中的一个或多个,这些途径本身可以进一步组织成三个核心细胞过程:
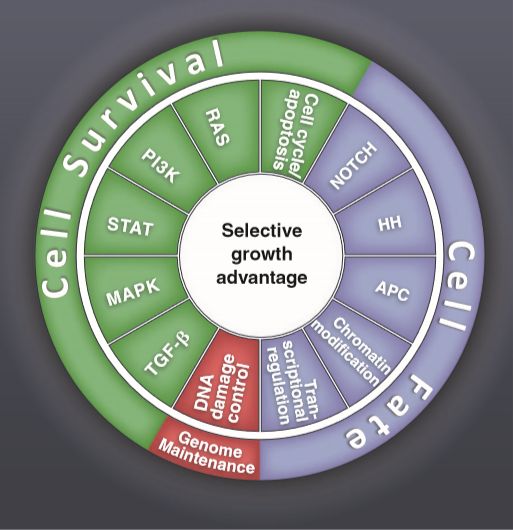
①细胞命运(Cell fate):大量研究表明细胞分裂与分化的关系是相反的,有细胞命运决定。负责填充正常组织的分裂细胞(干细胞)不能分化,反之亦然。再生医学是基于这种区别,以分化细胞去分化成干细胞,然后迫使干细胞分化成有用的细胞类型,以便移植回患者体内。癌症中的许多遗传改变取消了分化和分裂之间的精确平衡,且有利于后者。这会导致选择性生长优势,因为分化细胞最终会死亡或变为静止。通过这一过程起作用的途径包括 APC、HH和NOTCH,所有这些都是众所周知的,可以控制从蠕虫到哺乳动物等生物的细胞命运。 编码染色质修饰酶的基因也可以包括在这一类中。在正常发育过程中,从分裂到分化的可遗传性转换不是由突变决定的,而是由影响DNA和染色质蛋白质的表观遗传改变决定的。有什么比削弱表观遗传修饰装置本身更好的方法来破坏这种控制组织结构的正常机制呢?
NOTCH参考
②细胞存活(Cell survival):虽然癌细胞因如控制细胞命运的细胞的自主变化而异常分裂,但其周围的基质细胞完全正常。这种不对称最明显的分支是肿瘤的异常血管系统。与控制正常组织中营养素浓度的有序的动脉、静脉和淋巴网络不同,癌症的血管系统是曲折的,缺乏结构的一致性。正常细胞总是在离毛细血管100毫米的范围内,但对于癌细胞来说并非如此。因此,获得突变的癌细胞在限制营养浓度下增殖将具有选择性生长优势,在其姊妹细胞不能生长的环境中生长。例如,这种突变发生在EGFR、HER2、FGFR2、PDGFR、TGFBR2、MET、KIT、RAS、RAF、PIK3CA和PTEN基因中。其中一些基因编码生长因子自身的受体,而另一些基因则将生长因子的信号传递到细胞内部,激活后刺激生长。例如,KRAS或BRAF基因的突变赋予癌细胞以低于正常细胞或这些基因没有突变的癌细胞生长所需的葡萄糖浓度生长的能力。细胞周期的进展(及其对立面,细胞凋亡)可直接由细胞内代谢物控制,而直接调节细胞周期或细胞凋亡的驱动基因,如DCKN2A、MYC和BCL2,在癌症中经常发生突变。另一个突变能提高细胞存活率的基因是VHL,其产物通过血管内皮生长因子的分泌刺激血管生成。
③基因组维稳(Genome maintenance) :由于其所处的外来微环境,癌细胞暴露于多种有毒物质,如活性氧。即使没有微环境毒物,细胞在DNA复制或分裂过程中也会出现错误,而且在这种情况下,也会存在检测点来减慢这些细胞的速度或使它们自杀(凋亡)。虽然去除这些受损细胞对机体是有益的,但肿瘤细胞具有选择性生长优势可以在损伤中存活。如TP53和ATM基因的突变废除了这些检测点,这在癌症中并不奇怪。这些基因的缺陷可以间接地赋予选择性的生长优势,使具有有利于生长的总染色体变化的细胞(如易位或额外染色体)存活和分裂。类似地,控制点突变率的基因,如MLH1或MSH2,在癌症或易患癌症的患者的生殖系中发生突变,因为它们加速了调节细胞命运或存活过程的突变的获得。即增加驱动这个过程的突变的发生率来促进癌症。
由于调控细胞命运、细胞存活和基因组维稳的基因的蛋白质产物经常相互作用,它们之间的路径重叠;它们不像从上述描述中推断的那样离散。几个不同的基因可以为癌细胞产生相同的选择性生长优势,并且这些基因的产物相互作用。
认识到这些途径对我们理解患者间异质性的能力也有重要影响。一种肺癌可能在刺激性生长因子的受体中有激活突变,使其能够在低浓度的表皮生长因子(EGF)中生长。第二种肺癌可能在KRAS中有激活突变,KRAS的蛋白质产物通常将表皮生长因子受体(EGFR)的信号传递给其他细胞信号分子。第三种肺癌可能在NF1(一种通常使KRAS蛋白失活的调节蛋白)中有失活突变。最后,第四个肺癌可能在BRAF中发生突变,BRAF将信号从KRAS传递到下游激酶。单一通路中不同成分的突变是相互排斥的,也就是说,不发生在同一个肿瘤中,这已经被实验证实。
十、A Perspective on Genome-Based Medicine in Oncology
Opportunities
认识到某些肿瘤含有编码蛋白激酶的驱动基因的激活突变,导致针对这些激酶的小分子抑制剂药物的开发。这类基于基因组的药物的代表性例子包括使用EGFR激酶抑制剂治疗EGFR基因突变的癌症,前述的间变性淋巴瘤激酶(ALK)抑制剂治疗ALK基因易位的癌症,以及突变BRAF的特异性抑制剂治疗BRAF突变的癌症。在使用这种药物进行治疗之前,必须确定癌症是否含有药物靶向的突变。只有一小部分肺癌患者有EGFR基因突变或ALK基因易位,只有这些患者会对药物产生反应。而治疗没有这些特殊的基因改变的肺癌患者将是有害的,因为这些患者在肿瘤进展时会产生药物的毒性副作用。
第二种基于基因组的药物侧重于治疗药物的副作用和代谢,而不是针对的基因改变。目前,给病人服用的癌症药物的剂量是根据病人的体型(体重或体表面积)而定的。但是癌症药物的治疗率(导致副作用的浓度与杀死肿瘤细胞所需浓度的比值)通常很低,特别是对于传统(非靶向)治疗药物。这些药物循环浓度的微小变化可使肿瘤实质性消退与不可耐受的副作用产生差异。查询编码药物代谢酶的基因的种系状态可以通过加大给药显著改善治疗结果。
Challenges
所有针对基因改变产物的临床批准药物都是针对激酶的。其中一个原因是激酶相对容易以小分子靶向定位,并且在生物化学、结构和生理学水平上已被广泛研究。目前市场上绝大多数用于治疗癌症或其他疾病的药物都会抑制其蛋白质靶点的作用。这种抑制是因为药物干扰蛋白质的酶活性(如激酶催化的磷酸化)或蛋白质与小配体(如G蛋白偶联受体)的结合。但只有少部分原癌基因具有以这种方式靶向的酶活性。其他许多参与蛋白质复合物,涉及大界面和许多弱相互作用。因为小化合物只能抑制其中一种相互作用,所以使用小药物抑制这种蛋白质的功能非常的困难。
G蛋白偶联受体参考
大部分“Mut-driver”基因编码抑癌基因。药物通常会干扰蛋白质功能;一般来说,它们不能取代缺陷基因的功能,如由抑癌基因基因突变引起的功能。不幸的是,**在最常见的实体肿瘤中,相比原癌基因激活突变,抑癌基因基因失活突变占主导地位:很少个别肿瘤含有一个以上的原癌基因突变。 **
鉴于先前所述的转移内异质性,肿瘤中相对较少的癌基因突变是重要的。为了避免对靶向治疗产生不可避免的耐药性,可能需要用两种或两种以上的药物治疗患者。一个大转移病灶内的单个癌细胞对两种靶向两个独立通路的药物产生耐药性的概率是指数级小于该细胞对一种药物产生耐药性的概率。然而,如果癌细胞不包含一个以上的靶向基因改变(即原癌基因突变),那么这种组合策略是不可行的。
每一个肿瘤抑制基因失活都会导致通路下游一些促生长信号的激活。PTEN突变提供了一个例子:抑癌基因PTEN的失活导致AKT激酶的激活。同样,抑癌基因CDKN2A的失活也会导致诸如细胞周期蛋白依赖激酶4等促进细胞周期穿越的激酶的激活。此外,抑癌基因APC的失活导致原癌基因如CTNNB1和CMYC的组成活性。
对这些途径及其功能的研究有助于开发针对抑癌基因缺陷的药物,哪怕是间接的。事实上,已经有了这种间接瞄准的例子。抑癌基因BRCA1或BRCA2的失活突变导致在BRCA功能缺失的情况下激活修复DNA损伤所需的下游途径。因此,BRCA1或BRCA2缺陷的癌细胞更容易受到DNA损伤剂或抑制酶的药物的影响,这些酶有助于修复DNA损伤,如聚(腺苷二磷酸核糖)合酶PARP。PARP抑制剂在临床试验中显示了令人鼓舞的结果,用于肿瘤中有BRCA基因失活突变的患者。
通路功能是不同的,这取决于有机体、细胞类型和该细胞的精确遗传改变。用抑制突变BRAF激酶活性的药物治疗的结果提供了这一原理的一个相关例子。在大多数BRAF基因突变的黑色素瘤患者(V600E;V,Val;E,Glu)中,这些药物可显著(尽管短暂)缓解。但同样的药物对携带相同BRAF突变的结直肠癌患者没有治疗作用。这种观察被认为是由于EGFR的表达引起的,这种表达发生在一些结直肠癌中,但不发生在黑色素瘤中,并且被认为可以规避BRAF抑制剂的生长抑制作用。生物体是不同的,细胞类型通常是不同的,精确的基因构成总是不同的。一种在动物试验中失败的药物不一定在人类试验中失败,亦或相反,一种新的药物在小鼠的工程肿瘤中很有效,但可能在人类试验中却失败。
Genome-Based Medicines of the Future
真正的肿瘤特异性抗原可以被整合到许多已经存在的癌症免疫治疗平台中的任何一个。这些包括注射含有突变肽的疫苗、在其表面编码突变肽的病毒、呈递突变肽的树突状细胞以及针对突变肽具有反应性的抗体或T细胞。
要实现这类治疗,必须满足几个条件。首先,必须表达突变蛋白。由于癌细胞通常表达约一半由人类基因组编码的蛋白质,这种情况并不受限制。第二,由于大多数受突变影响的蛋白质是细胞内的,除非突变残基出现在人类白细胞抗原(HLA)蛋白质中,否则这些突变对免疫系统是不可见的。基于结合亲和性的生信分析,估计典型的乳腺癌或结直肠癌含有7到10个突变蛋白,这些突变蛋白可以结合个体患者的HLA蛋白。这些理论预测最近得到了实验支持。对小鼠肿瘤的研究已经确定了突变基因,并表明相应的肽在作为疫苗使用时能够诱导抗肿瘤免疫。此外,对脑癌患者进行免疫接种也获得令人鼓舞的结果。
如果一个肿瘤表达一种可以识别为外来的突变蛋白,为什么宿主免疫系统还没有根除这个肿瘤呢?事实上,肿瘤中的免疫编辑已经被证明是存在的,这导致了本该存在的突变表位发生下调或缺失,而突变表位本应在肿瘤发育过程中引起免疫应答。此外,肿瘤可以通过多种遗传改变失去免疫原性,从而阻止表位的出现,否则将被认为是外来的。
Other Ways to Reduce Morbidity and Mortality Through Knowledge of Cancer Genomics
建议“A计划”应该是预防和早期发现,“B计划”(晚期癌症治疗)应该只有在A计划失败时才有必要。为了使计划可行,政府和慈善组织必须将其大部分资源用于这一事业,并考虑到长期的考虑。我们相信,在未来几十年(152年),癌症死亡人数可以减少75%以上,但只有在为早期发现和预防作出更大努力的情况下,才能实现这一减少。