简介
热图是使用颜色来展示数值矩阵的图形,图中每一个小方格都是一个数值,按照预设的颜色值对应不同的数值。通常还会结合行、列的聚类分析,以表达实验数据多方面的结果。
热图在生物学领域应该广泛,尤其在高通量测序的结果展示中很流行,如样品-基因表达,样品-OTU相对丰度矩阵非常适合采用热图呈现。
在16s rDNA下游分析中,一般根据所有样本在属水平的物种注释及丰度信息,选取丰度排名前35的种属,从物种和样本两个层面进行聚类并绘制成热图,便于发现哪些物种在哪些样本中聚集较多或含量较低。
由于阅读数字时需要思考和比较,无法形成大范围的感官印象,而热图采用颜色的深浅代替数据表使得很多规律性的结果更加明显,而且热图在非常小的区域展示了大量的基因表达/细菌丰度数据,既可以快速比较组间的变化,同时还可以显示组内每个样品的的丰度,以及组内各样品间的重复情况,便于从中挖掘规律。结合聚类结果,使得整个实验的结果更加清晰和易于解释。
实例解读
例1. 热图展示在不同样品中的相对丰度1
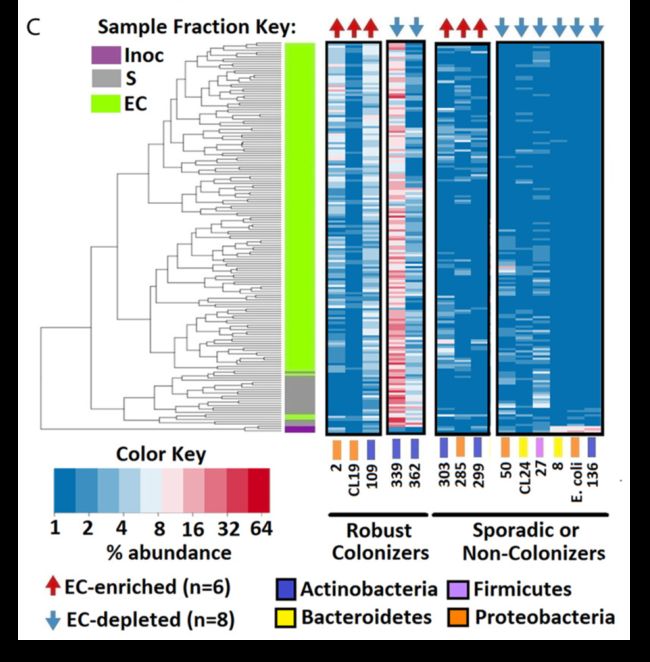
图1. 热图展示在不同样品中的相对丰度(Lebeis, S. L., et al.(2015)
该文是最早植物人工重组菌群的文章,研究了植物水杨酸对微生物组的影响,开山之作值得阅读。
-
图片元素解读
左侧聚类图为所有样品聚类的结果,左上角的图例代表三大类样品,紫、灰和绿它们分别代表接种菌、土壤和根样品,颜色标签在热图中第一列,用以区分样品组;
右侧为图的主图区,展示左侧样品中对应筛选的14个差异丰度菌的相对丰度值,丰度值百分比采用log2转换来缩小数据范围,并按从小到大对应的颜色梯度为蓝、白、红,即越红越高,越蓝越低。对应的图例为下方左上角的Color Key;
右侧正文区上方红上或蓝下箭头,代表这些菌的表达样式,为上调或下调,对应的图例为下方图例区的左下方(EC-enriched/depleted);
右侧正文区下方菌的标签上还有颜色,对应最下面图例区的菌门信息;同时菌还继续分为两类,稳定定殖者(Robust Colonizers)和偶然或非定殖者(Sporadic or Non-Colonizers)。
图表结果:图中展示了人工重组的菌在接种后,也可以形成丰度各异的微生物群体,并与自然条件下很多样式保持一致。
图表结论或规律:受水杨酸调控差异表达的菌,可以在人工重组实验中得到验证。
图片优点:配色采用红白蓝,比较严肃;图中添加了聚类信息、分组信息和菌分类信息,极大的增加了图片的信息和可读性。
例2. 免疫治疗反应与肠道微生物组成差异2
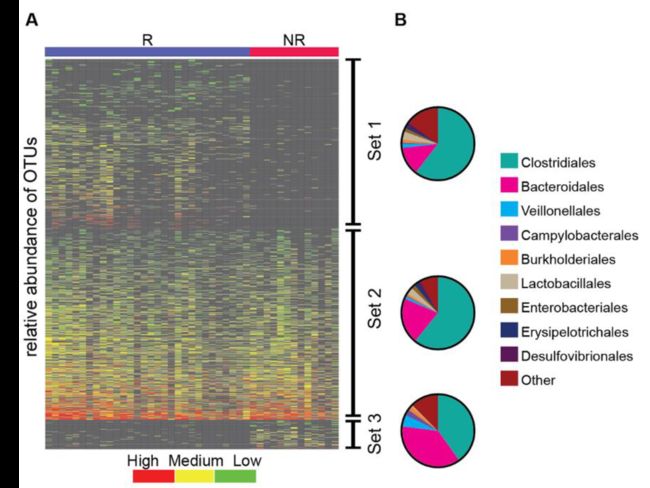
图2. 肠道微生物组成差异与免疫治疗反应相关(Gopalakrishnan Vancheswaran, et al.(2018)
-
图片元素解读
- 左上方展示了作者通过影像学评估对病人进行的分类responder(R) 和 noresponder(nr),用以表示样本的差异。
- 中间为图的主图区,展示上方对应样品中的OTU相对丰度值,按从小到大对应的颜色梯度为红、黄、绿,即越红越高,越绿越低。对应的图例为下方左上角的Color Key;
- 右侧线段,代表这些OTUs被区分为3类,set1(在R中富集),set2(不富集),set3(在NR中富集)。
- 最右侧通过扇形图进一步展示目水平中丰度前10的物种分布情况;同时,扇形图上下排布位置与set分类对应。
图表结果:图中展示了免疫治疗后,R和NR组微生物组成发生了改变;并通过扇形图具体指示了目水平上的具体分布比例(top10)。
图片优点:配色采用红黄绿,对比比较强烈;结合OTUs富集分组信息和分组信息,丰富了解读角度。扇形图的添加,把热图的模糊差异信息具现化,使得读者能够近距离、真正看到具体分布比例的变化情况。
例3. 展示时间序列的相对丰度变化3

图3. 时间序列的相对丰度(Zhang, J., et al.(2019)
- 文中的图片描述
值得注意的是,从幼苗阶段到生长后期与氮循环相关的OTUs的相对丰度有所提高,表明水稻可能积极地协调环境微生物群落以调节土壤养分以达到最佳生长状态。
Notably, the relative abundance of nitrogen cycle-related OTUs increased during the later stage of rice growth in the field compared to plants at the seedling stage (Fig. 3f,h and Supplementary Table 7), suggesting that rice plants might actively coordinate the environmental microbial community to modulate soil nutrients for optimal plant growth.
绘图实战
pheatmap 主要参数
pheatmap 包似乎只有pheatmap 一个函数,应该是用grid底层系统构建的,因此可以利用grid系统的相关函数进一步添加组分。由于只有一个函数,所有作者将所有的参数都压缩到了一起,可以通过?pheatmap查看,常用的有:
mat:用于可视化的数据矩阵
color: 配色要求
cellwidth/cellheight: 矩形色块的规格
treeheight_row/col:聚类树的规格
cluster_rows/cluster_cols:是否对或列聚类(TRUE/FALSE)
clustering_distance_rows/cols:聚类时使用的距离类型,和dist()函数相同
clustering_method:聚类方法,与hclust 相同
show_colnames/rownames: 是否显示行/列名(TRUE/FALSE)
annotation_row/col:给行或列添加注释信息
基础热图
# load package
library(pheatmap)
# set sample data
otu <- read.delim("otutab.txt", header = T, row.names = 1)
design <- data.frame(
samples = colnames(otu),
group = rep(c("KO", "WT", "OE" ),each = 6))
# head(otu)
# 按每个OTU的总丰度排序
otu <- as.data.frame(otu)
otu$sum <- rowSums(otu)
otu_order <- otu[order(otu$sum, decreasing = TRUE), ]
# head(otu_order)
# 取丰度前30的OTUs
mat <- otu_order[1:30, -ncol(otu)]
# create heatmap using pheatmap
pheatmap(mat = mat)

# 比较完整的参数
pheatmap(mat = mat,
border_color = 'grey60',
treeheight_col = 5,
cellwidth = 15, cellheight = 5,
cluster_row = F, cluster_col = T,
show_rownames = F, show_colnames = T,
clustering_method = "complete")

数据转换(归一化/标准化)
如果使用原始相对丰度或表达值,范围通常为0-100或0-1000000,而大部分的OTU或基因较低,做出的图会使绝大数据的数量颜色处于低丰度区,很难发现规律;因此需要数据变换,常用的方法有两类:
-
log2(x+1) x为丰度或表达值
给原始值+1是为了保证结果仍为正值,因为2的0次方为1;
为什么要使用log变换,以log2为例,0-1000的表达范围,经变化为0-10的范围,颜色梯度范围更容易使人与数值建立对应关系。
为什么常用log2对数变化,因为筛选差异的标准通常为两倍,log2对数变化后,每相差1的两个值都有两倍差异,选择目标很方便;有时也会根据具体情况,选择ln, log10等转换方式;
Z-score标准化:标准分数(standard score)也叫z分数(z-score),是一个分数与平均数的差再除以标准差的过程。用公式表示为:z=(x-μ)/σ。其中x为某一具体分数,μ为平均数,σ为标准差。 此种方法可以使有差异且稳定变化的两组明显区分为不同的颜色,但却丢失了原始相对丰度、差异倍数的信息。但由于结果比较美观,规律明显,使用较多。
# log2 转换
# scale_test <- apply(mat, 2, function(x) log2(x+1))
# scale 转换
scale_test <- apply(mat, 2, scale)
pheatmap(mat = scale_test,
# 边框颜色
border_color = 'grey60',
# 聚类树枝长度
treeheight_col = 5,
# cell 规格
cellwidth = 15, cellheight = 5,
# 是否进行聚类
cluster_row = F, cluster_col = T,
# 是否展示行列名
show_rownames = F, show_colnames = T,
# 聚类方法,与hclust相同
clustering_method = "complete")
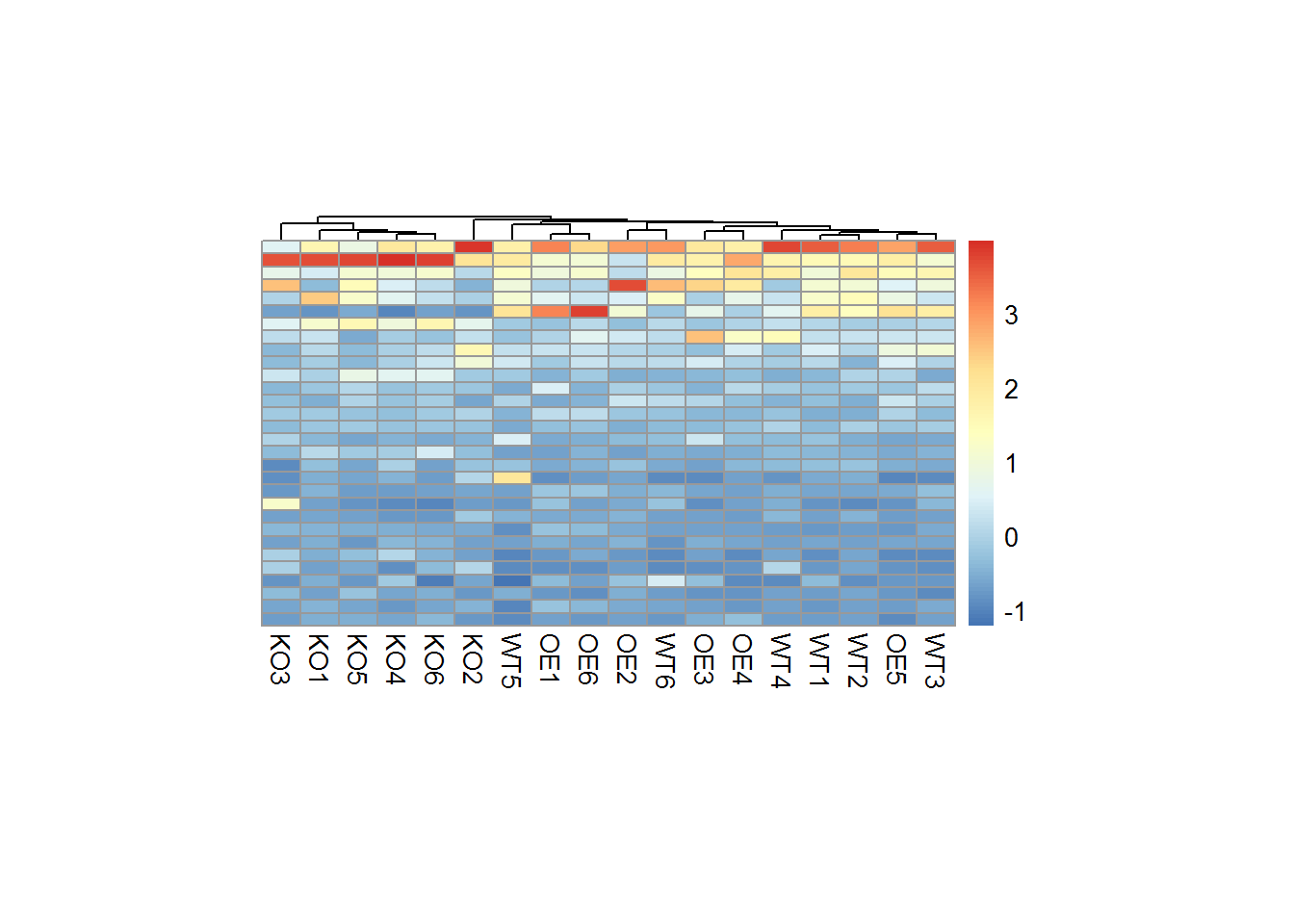
根据聚类结果分割热图
可以依据聚类簇将热图板块分开,这样我们就可以在热图主体中直接获得不同聚类簇的信息,而不会分心去查看聚类情况,在大量数据聚集在一起的时候,非常好用。
pheatmap(mat = scale_test,
# 边框颜色
border_color = 'grey60',
# 聚类树枝长度
treeheight_col = 5,
# cell 规格
cellwidth = 15, cellheight = 5,
# 是否进行聚类
cluster_row = F, cluster_col = T,
# 是否展示行列名
show_rownames = F, show_colnames = T,
# 聚类方法,与hclust相同
clustering_method = "complete",
# 分割热图
cutree_cols = 3)

热图色键分配
# color palette and cell size set
# divide the data range into several parts, say 50
# set color palette according to the divided data range
# 利用colorRampPalette 进行色键分割,参考?colorRampPalette
# 要求 col2rgb()
first <- colorRampPalette(c("white", "navy"))(10)
second <- colorRampPalette(c("navy", "red"))(40)
palette <- c(first, second)
pheatmap(mat = scale_test,
# 配色板
color = palette,
# 边框颜色
border_color = 'grey60',
# 聚类树枝长度
treeheight_col = 5,
# cell 规格
cellwidth = 15, cellheight = 5,
# 是否进行聚类
cluster_row = F, cluster_col = T,
# 是否展示行列名
show_rownames = F, show_colnames = T,
# 聚类方法,与hclust相同
clustering_method = "complete",
# 分割热图
cutree_cols = 3)

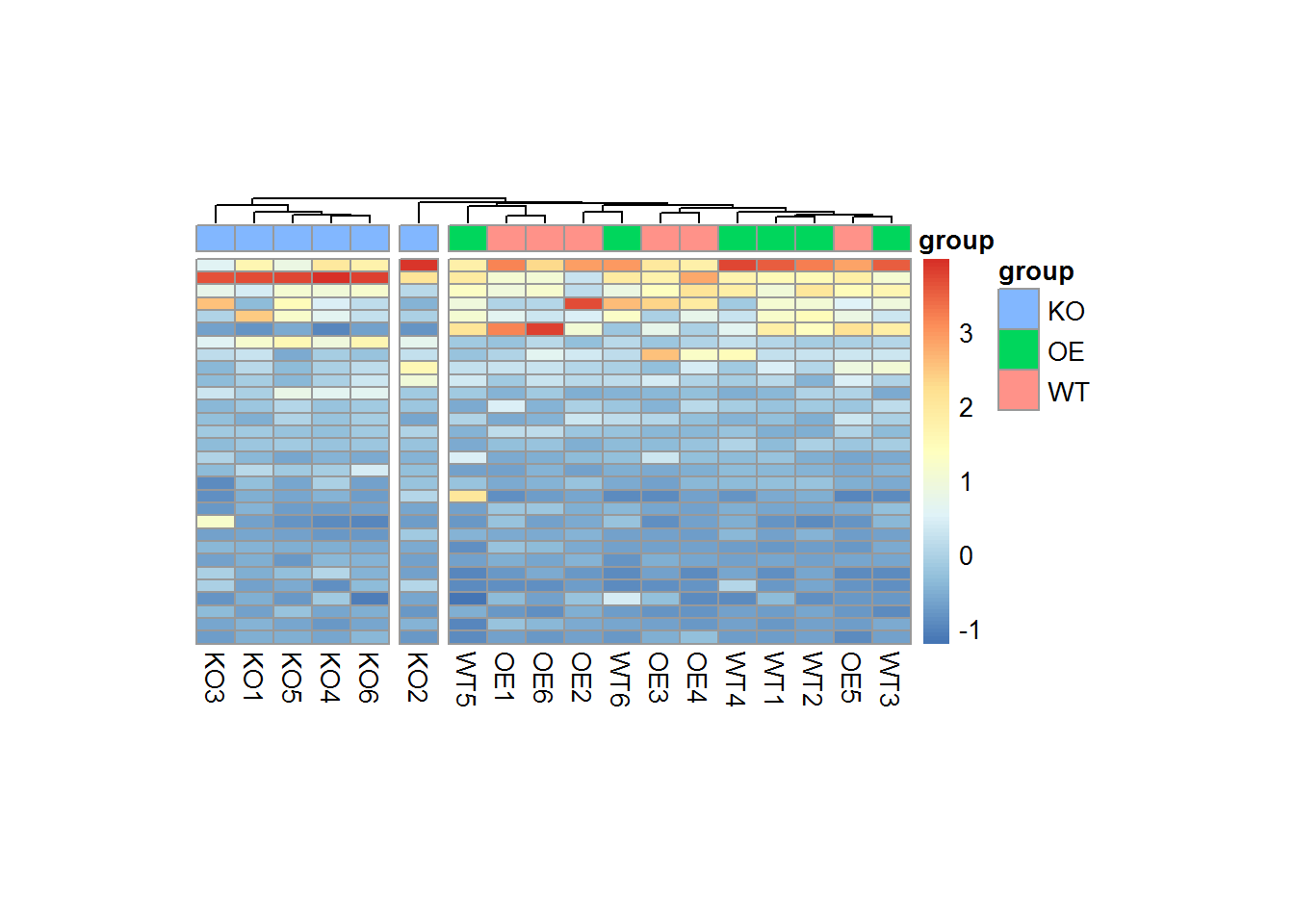
添加注释信息
# add annotation to heatmap
# add annotation data out of scratch
# 随机生成的分组x信息
# 如果分组信息是一个表格,则用merge()依据sample 合并即可
annot_data <- data.frame(row.names = colnames(mat),
group = design$group)
# head(annot_data)
pheatmap(mat = scale_test,
# 聚类树枝长度
treeheight_col = 5,
# cell 规格
cellwidth = 15, cellheight = 5,
# 是否进行聚类
cluster_row = F, cluster_col = T,
# 是否展示行列名
show_rownames = F, show_colnames = T,
# 聚类方法,与hclust相同
clustering_method = "complete",
# 分割热图
cutree_cols = 3,
# 添加注释
annotation_col = annot_data)
热图保存
save_pheatmap_pdf <- function(x, filename, width = 11,
height = 7) {
print("The unit of width and height are inches")
pdf(filename, width = width, height = height)
grid::grid.newpage()
grid::grid.draw(x$gtable)
dev.off()
}
#save_pheatmap_pdf(first_heatmap,
# filename = paste0(file,
# "water_genes_heatmap.pdf."))
Reference
1. Lebeis, S. L. et al. Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa. Science 349, 860–864 (2015).
2. Gopalakrishnan, V. et al. Gut microbiome modulates response to anti–pd-1 immunotherapy in melanoma patients. 359, 97–103 (2018).
3. Zhang, J. et al. NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice. Nat Biotechnol 37, 676–684 (2019).