aPRBind:结合序列和基于卷积神经网络学习的基于I-TASSER模型的结构特征,预测蛋白质-RNA相互作用
目录
-
- Paper_Info
- 预备知识:
- Abstract
- Motivation:
- Results:
- 1 Introduction
- 2 Methods
-
- 2.1 Data
- 2.2 基于 I-TASSER的结构构件
- 2.3 Features extraction
- 2.4 Convolutional neural networks
- 2.5 aPRBind算法的体系结构
- 2.6 Performance evaluation measures
- 3 Results
-
- 3.1采用I-TASTER搭建的结构
- 3.2 Analyses of feature contributions
- 3.3 aPRBind在独立测试集上的性能及其与现有预测服务器的比较
- 3.4 I-tasser模型精度对结合位点预测的影响
- 3.5 Case study
- 4 Conclusions
Paper_Info
作者:Yang Liu, Weikang Gong, Yanpeng Zhao, Xueqing Deng, Shan Zhang, and Chunhua Li
单位:北京工业大学环境与生命科学学院
发布期刊:Bioinformatics
发布时间: 2020年8月21日
paper
code
dataset RB198、RB111
补充材料
预备知识:
aPRBind是一种预测蛋白质上RNA结合残基的方法。
I-TASSER(Iterative Threading ASSEmbly Refinement):I-TASSER(迭代线程优化)是一种用于蛋白质结构预测和基于结构的功能注释的分层方法。它首先通过多线程方法LOMETS从PDB识别结构模板 ,并通过基于迭代模板的片段装配仿真构建全长原子模型。然后,通过蛋白质功能数据库BioLiP重新对3D模型进行穿线,从而得出目标的功能见解 。I-TASSER(作为“ Zhang-Server”)在最近的社区范围内的 CASP7, CASP8, CASP9, CASP10, CASP11, CASP12, CASP13和 CASP14 实验。它在CASP9中也被认为是功能预测最好的 。
输入为:FASTA格式序列[10,1500]个残基内 。
输出:蛋白质结构和功能预测。
I-TASSER网站
AA指数获取理化性质特征
步骤:
包括三个步骤:
1.I-TASSER模型构建
https://zhanglab.ccmb.med.umich.edu/I-TASSER/,to construct the protein structure and the first model (named as 2l5d_a.pdb) out of the five predicted ones is selected. Parameters are set to default values.
2.The second step: Features extraction
,特征提取和预测性能。
Abstract
Motivation:
蛋白质-RNA相互作用在各种生物过程中起着至关重要的作用。蛋白质中RNA结合残基的准确预测一直是计算生物学领域中最具挑战性和最具吸引力的问题之一。现有的方法仍然具有相对较低的精度,特别是对于基于序列的ab-initio方法。
Results:
在这项工作中,我们提出了一种基于卷积神经网络(CNN)的ab-initio 方法,用于RNA结合残基的预测。APRBind用从I-tasser预测结构中提取的序列特征和结构特征(特别是包括我们开发的残基动力学信息和残基核苷酸倾向)进行训练。特征贡献率分析表明,序列特征在结合位点预测中最重要,其次是动力学信息,序列特征和结构特征是互补的。在基准数据集上与其他同类方法的性能比较表明,aPRBind方法的性能优于一些先进的ab-initio方法。此外,由于结构特征对精化的三维结构不是很敏感,aPRBind对结构模型的精度只有很小的依赖关系,这使得aPRBind可以应用于对建模(对于TM-Score≥0.5的建模结构)或未绑定结构的RNBind位点预测。
Availability: 源代码可在https://github.com/ChunhuaLiLab/aPRbind.上获得
1 Introduction
蛋白质-RNA相互作用在广泛的生物学过程中发挥着关键作用,如基因表达和调控、蛋白质合成和病毒组装(Keene,2007)。蛋白质-RNA识别的异常可能导致许多疾病(LuKong等人,2008年)。因此,蛋白质上RNA结合残基的可靠识别是一个重要而又具有挑战性的问题,它对于理解蛋白质-RNA相互作用的识别机制至关重要,也有助于复杂结构预测和药物设计。实验方法非常昂贵和耗时。因此,开发预测蛋白质-RNA结合位点的计算方法的需求越来越大。在过去的十年中,已经建立了许多计算方法。根据它们使用的特征,这些方法可以分为两类:基于序列的方法(Carson等人,2010;El-Manzalawy等人,2016;Kumar等人,2008;Murakami等人,2010;Terribilini等人,2007;Walia等人,2014)和基于结构的方法(Chen和Lim,2008;Kim等人,2006;Maetschke和袁,2009年;Perez-Cano和Fernandez-Recio,2010年;唐等人,2017年;Towfi等人,2010年)。
对于基于序列的方法,目标蛋白的常用特征包括位置特定评分矩阵(PSSM)、氨基酸理化性质、预测的溶剂可及性等。PSSM是序列进化特征的一种常见代表,由于界面残基经历了相对较高的进化压力,比其他表面残基发生的突变更少(更保守),因此PSSM已被广泛应用于大多数预测因子中。除了标准的PSSM外,还开发了两种主要的改进的PSSM图谱,并将其用于RNA结合残基预测,这两种图谱分别通过滑动窗口(El-Manzalawy等人,2016;Li等人,2014;Walia等人,2014)和平滑处理后的滑动窗口(称为平滑PSSM)(Cheng等人,2008年)进行缩放。与平滑处理和滑动窗口均基于目标残差序列邻域的平滑PSSM不同,本文提出的SNB-PSSM(Space Neighbor Based Position-Specific Score Matrix)采用基于空间邻域的平滑处理和结构窗方案对进化信息进行编码。SNB-PSSM比平滑的PSSM对RNA结合残基的预测更好,这在一定程度上可以用我们检测到的保守的界面残基经常聚集在一起来解释(Yang等人,2020年)。对于基于结构的方法,除了序列特征外,常用的结构派生特征包括结构特征和拓扑特征等。前者主要包括二级结构类型、几何裂隙(Chen和Lim,2008)、复杂网络性质(Maetschke和袁,2009)、Voronoi接触和结构邻居(唐等,2017),后者主要包括溶剂可及性(Maetschke和袁,2009)和静电势(Chen和Lim,2008)。
除了上述特征外,还需要探索其他一些特征来推进RNA结合位点的预测。众所周知,蛋白质RNA相互作用具有序列和结构识别特异性(Chen等人,2004年;Jeong等人,2003年;Jones等人,2001年;Perez-Cano和Fernandez-Recio,2010年)。基于这一事实,我们提取了60×8个残基-核苷酸成对倾向势,其中考虑了先前工作中的二级结构信息,这在区分近天然复杂结构方面表现出了良好的性能(Li等人,2012年)。结合物理能项,这一势可以捕捉到评分列表前5名中至少有一种对接模式,该模式在91.4%的蛋白质-RNA相互作用的界面上天然界面残基和核苷酸不低于50%(Zhang等人,2017年),显示出良好的界面预测能力。因此,在这里,我们尝试将其作为一个特征应用于RNA结合残基的预测。此外,除了序列和结构特征外,蛋白质动力学特性在蛋白质-蛋白质/配体特异性识别和相互作用中发挥着重要作用,这些特性已经被用来预测结合关键残基、结合热点(Melo等人,2016),甚至变构位点(Taguchi和Kitao,2016)。我们还利用残基动力学特性成功地确定了snRNA与人U1A蛋白相互作用的关键结合残基(han等人,2019年)。因此,残基动力学属性也是我们想要应用于RNA结合位点预测的。
近年来,随着蛋白质结构预测方法的发展,在没有同源模板的情况下具有很高的预测精度,这使得我们能够构建基于预测结构的结合位点预测方法。张等人开发的I-Tasser结构预测器是一种基于线程的方法,即使对于新的折叠目标也表现得相当好,在过去十年的全社区CASP(蛋白质结构预测关键评估)实验中一直处于领先地位(Yang等人,2015年)。
在这项工作中,我们提出了一个基于序列的从头算法aPRBind(ab-initio Protein-RNA Binding Site Forecast)来预测蛋白质中的RNA结合残基,该算法利用了基于SNB-PSSM的序列特征和基于I-tasser模型的结构特征(包括残基动力学特性和残基核苷酸倾向)。这些特征是通过深度卷积神经网络模型学习的。
2 Methods
2.1 Data
在这项工作中,我们使用基准数据集RB198(El-Manzalawy等人,2016)作为训练集。RB198中的数据是通过从PDB中的蛋白质-RNA复合物中去除满足以下任何标准的复合物而得到的:i)结构分辨率低于3.5µ;ii)蛋白质残基<40或RNA核苷酸<5;iii)界面残基<3;iv)与其他链的序列同源性>30%。RB198数据集有134个复合体,198个蛋白链。
El-Manzalawy等人对许多蛋白质-RNA界面预测服务器进行了相互比较。(El-Manzalawy等人,2016)在基准数据集RB111上,我们使用RB111作为独立的验证数据集,将我们的方法与其他服务器进行比较。
对于这两个数据集,蛋白质的界面残基被定义为至少有一个原子与其伙伴RNA的任何一个原子的距离大于5 A 。 A{^。} A。根据它们是结合残基还是非结合残基,所有残基都被标记为“1”或“0”。在RB198数据集中有7878个结合残基,43150个非结合残基,在RB111数据集中有为3305个结合残基和34255个非结合残基。
2.2 基于 I-TASSER的结构构件
I-TASSER,一种基于线索的蛋白质结构预测方法,由Zhang等人提出。(Yang等人,2015)被用来从它们的序列中构建蛋白质结构。它通过迭代重组从线程模板中提取的结构片段来分层构建全长模型。参数设置为默认值。另外,需要指出的是,在蛋白质结构构建中,所有与查询序列同源性 >30%的模板都被排除在模板库之外。最后,从五个模型中选出TM得分最高的第一个模型作为构建结构。
2.3 Features extraction
我们使用的特征包括序列特征和结构特征。除了基于SPIDER3的特征和物理化学性质外,以下所有特征都属于由I-TASSER从建模结构中提取的结构特征。
基于空间邻居的特定位置评分矩阵(SNBPSSM):
考虑到保守的界面残基经常聚集在蛋白质三级结构中(Ahmad等人,2008;Capra和Singh,2007;Guharoy和Chakrabarti,2010),我们在以前的工作中提出了一种新的进化信息编码方案,即基于空间邻居的PSSM(SNB-PSSM),它不同于平滑的PSSM(Cheng等人,2008)。
在SNB-PSSM方法中,首先,对于含有N个残基的蛋白质,PSSM矩阵的大小为20×N,每个位置都有进化信息。然后,进行基于空间邻域的平滑处理,使目标残基的进化分数为C-α原子与目标残基的进化分数之和的平均值。最后,采用基于空间邻域的窗口方案,将目标残留物的进化信息编码为与目标残留物位置空间最近的25个残留物位置的平滑进化分数。因此,对于一个目标残基,它的进化信息被编码到一个20×25的矩阵中。该编码过程考虑了目标残基周围的空间邻居的演化。
接口倾向(IP):我们从251个非冗余的蛋白质-RNA复合物中提取了考虑蛋白质和RNA二级结构信息的残基-核苷酸倾向性(60×8)作为特征(Li等人,2012)。在倾向性上,蛋白质和RNA的二级结构根据它们的界面倾向性分别被分为三类和两类。特定残基-核苷酸对的倾向性是通过其在界面上的观察概率除以其预期概率来计算的。这里,具有某一类二级结构的残基类型的界面倾向被表示为其对8种核苷酸的成对倾向的平均值。
残留物波动动态:残基波动动力学的特征是由高斯网络模型(GNM)计算的,该模型基于谐波势,已被证明是再现生物大分子内在动力学的可靠方法(Bahar等人,1997)。N个节点的网络的总内部势能可以写为:
V = 1 2 γ [ Δ R T ( Γ ⊗ E ) Δ R ] V=\frac{1}{2} \gamma\left[\Delta \boldsymbol{R}^{T}(\Gamma \otimes \boldsymbol{E}) \Delta \boldsymbol{R}\right] V=21γ[ΔRT(Γ⊗E)ΔR]
γ是弹簧的简谐力常数,列向量∆R表示N个节点的涨落,上标T表示转置,E是3×3恒等式矩阵, ⊗ \otimes ⊗是矩阵直积, Γ \Gamma Γ是N×N对称基尔霍夫矩阵。第i残基的均方起伏可以表示为:
⟨ Δ R i ⋅ Δ R i ⟩ = 3 k B T γ [ Γ − 1 ] i i ∝ ∑ k = 2 N λ k − 1 [ u k ] i 2 \left\langle\Delta R_{i} \cdot \Delta R_{i}\right\rangle=\frac{3 k_{B} T}{\gamma}\left[\Gamma^{-1}\right]_{i i} \propto \sum_{k=2}^{N} \lambda_{k}^{-1}\left[u_{k}\right]_{i}^{2} ⟨ΔRi⋅ΔRi⟩=γ3kBT[Γ−1]ii∝k=2∑Nλk−1[uk]i2
其中kB是玻耳兹曼常数,T是绝对温度。在GNM中,由于慢模式对残基波动的贡献最大(Bahar等人,1998年),我们计算了前m(1-6)个运动模式的相对残差波动(忽略3kBT/γ),作为残基波动特征表示为F1到F6。
基于SPIDER3的特征:程序SPIDER3用于从序列预测蛋白质二级结构(SS)状态和溶剂可及性(SA)(Heffernan等人,2017年)。此外,根据该序列计算的特征包括主链扭转角(φ和ψ)、Cα原子之间的主链角(θ和τ)以及根据每个考虑的残基的相邻Cα-Cα矢量定义的上半球和下半球内的半球曝光(Hseα_Up和Hseα_Down)。
深度和突出指数(DPX和CX):
采用PSAIA程序(Mihel等人,2008年)获得残留物深度和突起指数。原子的深度指数(DPX)被定义为它到最近的溶剂可及原子的距离。凸起指数(CX)由Vext/Vint定义,其中Vint是以所考虑的原子为中心的固定半径(这里为10?)球体内非氢原子占据的体积,球体的剩余体积为Vext。对于残基,它的DPX和CX分别是其所有非氢原子上DPX和CX的平均值。
拓扑特征:利用Python编程包NetworkX(v1.11)计算了每个残基的拓扑特征,包括度、聚类系数、贴近度中心度、介数和度中心度。这里,C-α原子被视为节点,如果两个节点小于7.5A,则两个节点由一条边连接。
理化性质:每种残基类型的10个物理化学性质是从AA指数数据库(Kawashima等人,2008年)中获得的,其中包括原子数量、静电电荷数量、潜在氢键数量、分子质量(M质量)、疏水性、亲水性、极性、极化率、倾向性和平均可获得表面积。此外,根据氨基酸的电荷类型,将其分为正电荷、负电荷和非电荷三个基团,分别用1、-1和0表示。
2.4 Convolutional neural networks
卷积神经网络(CNN)模型(Defferrard等人,2016)在TensorFlow(Rampasek和Goldenberg,2016)中实现,它包含两个卷积、两个最大汇集和两个完全连通的层。采用二维CNN结构。残差由24×24维的特征向量描述(SNB-PSSM为500维,其他特征为36维,零值为40)。这两个卷积层分别具有8个和16个滤波器,每个滤波器(核)大小为2×2。大小为2的最大池适用于这两个卷积层。因此,两个完全连接的层具有576个和64个单元。使用参数化整流线性单元(PReLU)作为激活函数,并将丢失率设置为0.5。对于真标签和采样的负类,交叉熵损失最小。我们使用50k迭代,小批量大小为100。
2.5 aPRBind算法的体系结构
APRBind的流程图如图1所示。查询的蛋白质序列被提交给两个程序PSI-BLAST和SPIDER3,以生成PSSM简档和基于SPIDER3的特征。此外,将该序列馈入I-Tasser Suite(Yang等人,2015年)以生成结构模型。然后基于所构建的结构模型计算了SNB-PSSM剖面、界面倾向、拓扑和动力学特性等结构特征。利用卷积神经网络在RB198训练集上进行aPRBind预测。
图1.RNA结合位点预测的aPRBind流程图。所使用的特征来自序列和I-tasser模型,这些模型是用深度卷积神经网络模型学习的。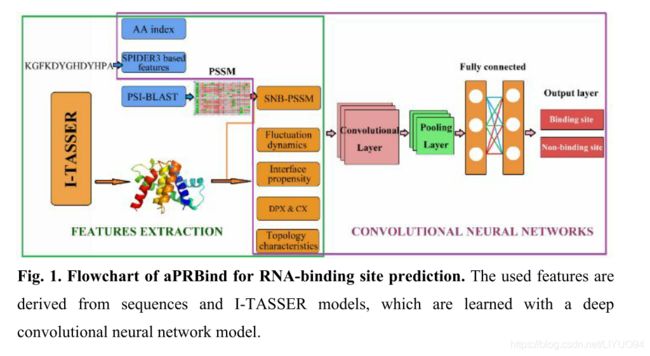
2.6 Performance evaluation measures
通过5次交叉验证对CNN模型进行训练,并采用独立数据集检验分类器的有效性。分类器的预测性能用定义如下的总体准确度(ACC)、灵敏度(SN)、特异度(SP)和马修斯相关系数(MCC)进行评估
A C C = T P + T N T P + F P + T N + F N S N = T P T P + F N S P = T N T N + F P M C C = T P × T N − F P × F N ( T N + F N ) ( T N + F P ) ( T P + F N ) ( T P + F P ) \begin{aligned} &A C C=\frac{T P+T N}{T P+F P+T N+F N}\\ &S N=\frac{T P}{T P+F N}\\ &S P=\frac{T N}{T N+F P}\\ &M C C=\frac{T P \times T N-F P \times F N}{\sqrt{(T N+F N)(T N+F P)(T P+F N)(T P+F P)}} \end{aligned} ACC=TP+FP+TN+FNTP+TNSN=TP+FNTPSP=TN+FPTNMCC=(TN+FN)(TN+FP)(TP+FN)(TP+FP)TP×TN−FP×FN
其中真阳性(TP)、假阳性(FP)、真阴性(TN)和假阴性(FN)是通过将每个残基的预测标签与实际标签进行比较而获得的。
3 Results
3.1采用I-TASTER搭建的结构
使用I-Tasser程序对RB198和RB111数据集中的蛋白质序列建立原子结构模型。ITASSER模型的准确性通过模板建模得分(TM-SCORE)(徐和张,2010年)进行评估,范围在0到1之间。通常情况下,TM-SCORE≥0.5的I-TASSER模型被认为是正确的折叠。
图S1显示了RB198和RB111数据集中309条蛋白链的建模结构的TM分值分布。从图S1看,大多数模型(82.5%)的TM得分在0.5以上,TM得分>0.8的模型占所有模型的21.4%。309个模型的TM得分平均值为0.65。结果表明,尽管所有与靶序列同源性 >30%的同源模板都被排除在模板库之外,但大多数I-tasser模型都有正确的折叠。
3.2 Analyses of feature contributions
使用最大相关最小冗余度(MRMR)(Peng等人,2005年)(见补充资料)来评估特征在RNA结合残基预测中的重要性。由于残基进化信息已被证明是许多预测因子中最具区分性和最有效的特征(El-Manzalawy等人,2016年;Kumar等人,2008年;Li等人,2014年;Walia等人,2014年),这里我们评估了其他特征的重要性。表S1给出了通过在RBP198数据集上应用mRMR获得的前20个特征,以及它们在界面和非界面残基上的平均值以及P值。
从表S1可以看出,界面残留物上的相对溶剂可及性(RSA)平均值比非界面残留物上的高45.1%,这意味着RSA值高的残留物很可能出现在界面上。界面残渣上的P(H)平均值比非界面残渣上的P(H)平均值低20.5%。我们以前的工作发现,α-螺旋中的残基不喜欢出现在RNA结合界面,这在一定程度上与目前的观察结果一致。(Li等人,2012年)。氨基酸的电荷类型排在第三位。由于核苷酸是带负电荷的,静电相互作用在蛋白质-RNA相互作用中发挥着重要作用,蛋白质-RNA相互作用中的正电荷氨基酸更喜欢与RNA分子结合(Li等人,2012年)。在这里,我们还计算了RB198和RB111数据集中每种氨基酸的界面(相对于非界面)倾向。结果如图S2所示。从图S2可以看出,氨基酸Arg、His和Lys的倾向性最高,分别为1.94、1.41和1.38,说明带正电的氨基酸更喜欢与RNA分子结合。此外,前m(1-6)运动模式(F1~F6)贡献的相对残基波动均在前14位,界面残基的平均相对波动至少比非界面的高146.7%,说明界面残基总体上具有较高的弹性。界面残留物的平均α_up比非界面残留物的低16.4%。界面残留物的平均IP值高于非界面残留物的平均IP值,这与IP本身的定义是一致的。平均深度指数(DPX)排在前10位,平均而言,界面残留物的DPX值比非界面残留物低。综上所述,除了基于序列的特征外,结构和动力学特征在RNA结合残基预测中起着重要作用。

表一:在RB198上考虑不同特征组合的CNN模型的5次交叉验证实验的平均结果。

此外,为了检测特征对CNN模型的贡献,我们使用考虑了不同特征组合的CNN模型进行预测,并对预测结果进行了比较。由于进化信息已被证明是许多预测因子中最具区分性和最有效的特征(El-Manzalawy等人,2016;Kumar等人,2008年;Li等人,2014;Walia等人,2014),所有模型都考虑了基于SNB-PSSM的特征。其他36个特征被分为六类:两类基于序列的特征(包括AA索引和基于SPIDER3的特征)和四类基于结构的特征(包括CX/DPX、IP、拓扑和动力学特征)。表1显示了不同CNN模型在RB198上的5次交叉验证实验的平均结果。对于基于SNB-PSSM的特征与不同单一特征类型的组合,考虑基于SPIDER3的特征的MCC值最好,其次是分别考虑AA指数、动力学、IP、拓扑和CX/DPX特征的特征。因此,基于序列的特征相对更重要,另外,从结构中获得的动力学特征也很关键,这与mRMR的单一特征分析结果是一致的。当基于序列的特征分别组合成四种类型的基于结构的特征时,动力学特征的组合获得了最好的结果(MCC为0.40),这表明它们在结合位点预测方面具有互补性。此外,与除拓扑特征和动力学特征外的所有特征的性能比较都考虑到动力学特征相对比拓扑特征更重要。最后,综合考虑所有特征的性能达到最佳,SN、SP、ACC和MCC分别为0.65、0.82、0.74和0.48。
总体而言,上述结果表明,基于序列的特征对CNN模型的预测最为重要,而据我们所知首次应用于蛋白质-RNA结合位点预测的动力学特征也是至关重要的。基于序列和基于结构的特征在结合位点预测中是互补的。
表2.RB111上aPRBind与支持向量机和RF模型以及部分现有蛋白质-核糖核酸界面预测服务器的 比较

3.3 aPRBind在独立测试集上的性能及其与现有预测服务器的比较
我们在独立测试集RB111上测试了考虑所有特征的训练好的CNN模型,结果如表2所示。同时,为了检验CNN深度学习方法对预测性能的影响,我们在RB111上用相同的方法测试了训练好的支持向量机(SVM)和随机森林(RF)模型。此外,表2给出了四个基于序列的蛋白质-RNA界面预测服务器(FastRNABindR(ElManzalawy等人,2016)、RNABindR v2(Walia等人,2012)、BindN+(Wang等人,2010)和PPRInt(Kumar等人,2008))在RB111数据集上的测试结果。从表2可以看出,支持向量机模型在所有评估指标上都略优于RF模型。我们的方法aPRBind得到的ACC、SN、SP和MCC分别为0.86、0.48、0.90和0.32,与SVM和RF模型相比,MCC分别提高了10.3%和18.5%。与FastRNABindR、RNABindR v2、BindN+和PPRInt四种预测服务器相比,aPRBind的ACC、SP和MCC值最高,MCC分别提高了33.3%、45.5%、33.3%和77.8%。
综上所述,在七种基于序列的ab-initio方法中,aPRBind在界面残基预测方面表现出较好的性能,我们认为这主要是由于考虑了基于结构的特征(如动力学特征、界面倾向和基于SNB-PSSM的进化信息)以及卷积神经网络算法的使用。
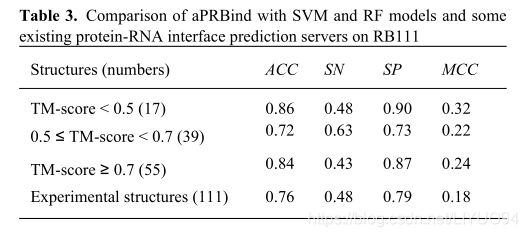
3.4 I-tasser模型精度对结合位点预测的影响
由于aPRBind使用的基于结构的特征是从ITASSER模型中提取的,所以我们想知道预测模型的质量是否会影响其预测性能。通常情况下,Tm-Score≥为0.5的ITASSER模型被认为是正确的褶皱。因此,为了发现问题,我们将所有模型分成三组,分别为TM-Score<0.5、0.5≤、TM-Score<0.7和TM-Score≥0.7。表3显示了aPRBind在这些组上的性能。从表3可以看出,TM-Score<0.5的组的表现比TM-Score≥为0.5的组的表现差。特别是TM-Score<0.3(2XLK:A和3T5N:A)的模型,其MCC值较低,分别为0.18和0.19。将后两组的表现与TM-SCORE≥0.5进行比较,可以看出TM-SCORE≥0.7组的表现稍好一些。此外,我们对Rb111的所有实验结构进行了aPRBind,结果也如表3所示。可以看出,aPRBind在实验结构上的性能与在TM-Score≥为0.7时的I-tasser模型上几乎相同,只是ACC值增加了0.01。
从以上结果可以得出结论,基于正确折叠的结构特征有助于aPRBind进行结合位点预测。另外,aPRBind在残基位置大致正确的情况下,对结构变化具有很好的鲁棒性,这主要是因为aPRBind使用的结构特征是粗粒度的,对精细的三维结构不是很敏感。
表3.RB111上aPRBind与支持向量机和RF模型以及部分现有蛋白质-核糖核酸界面预测服务器的 比较
3.5 Case study
图2显示了aPRBind对I-tasser模型Tm-Score<0.5和>0.5的两个蛋白质实例的预测结果,并给出了对其实验结构的预测结果以供比较。第一个例子是核衣壳蛋白四聚体(PDB ID:4H5P:A)(Raymond等人,2012年)。其i-tasser模型的TM得分为0.38。在I-tasser模型上,aPRBind的ACC、SN、SP和MCC分别为0.87、0.68、0.90和0.54,在实验结构上的相应结果分别为0.87、0.65、0.91和0.52(见图2(A)和(B))。
第二个例子是RBP,Human Disher Platform-PAZ-Connector Helix盒式磁带(PDB ID:4NGD:A)(Tian等人,2014)。i-tasser模型的TM-score 是0.53。在I-tasser模型上,aPRBind的ACC、SN、SP和MCC分别为0.91、0.68、0.93和0.52,在实验结构上的相应结果分别为0.91、0.77、0.92和0.55(见图2©和(D))。
从这两个实例可以看出,虽然i-tasser构造的结构不是很好,但aPRBind得到了比较满意的结果。对于4H5P:A,TM得分较低的主要原因是对突出螺旋的预测不好。因此,其I-tasser模型的预测结果与实验结构相似或略好。对于4NgD:A,I-tasser没有很好地预测界面的二级结构,但排列方式与实验结果相似。因此,对所建模型的预测结果仍然令人满意。

图2.I-tasser和实验求解的分别对建模结构上aPRBind与RNA结合残基的 预测。(A)和(B)PDB ID:4H5P:A的情况,ITASSER模型的结果分别为SN=0.68,SP=0.90,ACC=0.87,MCC=0.54(TM-SCORE=0.38),实验结构的结果分别为SN=0.65,SP=0.91,ACC=0.87,MCC=0.52。©和(D)PDB ID:4NGD:A,对于I-tasser模型(TM-Score=0.53),结果分别为SN=0.68,SP=0.93,ACC=0.91,MCC=0.52;对于实验结构,结果分别为SN=0.77,SP=0.92,ACC=0.91,MCC=0.55。蛋白质结构用灰色卡通图表示。TP、FP和FN结果分别以绿色、红色和蓝色显示。
4 Conclusions
我们提出了一种基于序列的从头算方法aPRBind(ab-initio Protein-RNA Binding Site Prediction)来预测蛋白质中的RNA结合残基,其中从I-tasser模型中提取的序列特征和结构特征是通过深度卷积神经网络模型来学习的。在结构预测中,为了满足一般情况,所有与靶序列一致性 >30%的同源模板都被排除在模板库之外。结果表明,虽然进行了上述处理,但大多数I-tasser模型(82.6%)具有正确的折叠,TM-Score不低于0.5。对特征贡献的分析表明,序列特征对预测的影响最大,动力学特征也很重要,序列特征和基于结构的特征在结合位点预测中是相辅相成的。APRBind在独立测试集上的性能表明,我们的方法可以对正确建模的褶皱给出更好的预测。另外,只要残基位置大致正确,aPRBind对结构变化具有很好的鲁棒性,这主要是因为aPRBind使用的结构特征是粗粒度的,对精细的三维结构不是很敏感。我们的方法优于SVM和RF模型,以及一些经典的基于序列的预测服务器FastRNABindR、RNABindR v2、BindN+和PPRInt。这项工作有助于加强我们对蛋白质-RNA识别和相互作用的理解,并可用于蛋白质-RNA对接预测和结合热点探索。