[toc]
1. 组装算法
一般有基于OLC(Overlap-Layout-Consensus, 先重叠后扩展)和基于DBG(De Brujin Graph)两种组装算法。基于OLC的组装方法适合长序列组装,运行依赖的数据结构需要消耗大量的内存,且运行速度比较慢,错误率高,而DBG组装方法内存消耗相对较低,运算速度快,且准确率高。目前主流的基因组装算法都是基于后者改进设计的。
1)基于OLC算法
OLC组装算法主要这么对一代和三代测序序列,因为它们的reads读长相对较长。OLC算法的整体步骤可以分为三步:

- ①Overlap:对所有reads进行两两比对,找到片段间的重叠信息,一般在比对之前会将reads做下索引,减少计算量。这里需要设定最小重叠长度,如果两个read的最小重叠长度低于一定阈值,那么可以认为两段序列顺序性较差。
- ②Layout:根据得到的重叠信息将存在的重叠片段建立一种组合关系,形成重叠群,即Contig。Contig进一步排列,生成多个较长的scaffold。
- ③Consensus:根据构成Contig的片段的原始质量数据,在重叠群中寻找一条质量最重的序列路径,并获得与路径对应的序列,即Consensus。通过Consensus的多序列比对算法,就可以获得最终的基因组序列。
OLC算法最初成功的用于Sange测序数据的组装,比如Celera Assembler,Phrap,Newbler等均采用该算法进行拼接组装。基于Overlap-layout算法的组装软件首推CABOG,这是当年用来组装果蝇基因组的原型。
2)基于DBG算法
DBG原理图如下,共6步:

A. 序列k-mer化:对插入片段进行建库测序,下机reads经质控后,对clean reads进行k-mer化,即将reads 逐个碱基开始切分为长度为K的子串;
B. 构建de Brujin图:将上一步得到的所有长度为k的子串即k-mer作为de Brujin图的节点,根据相邻两个K-mer重叠k-1个碱基的原则将该两个顶点(k-mer)有方向的连接起来,构建de Brujin图,如下图所示:

C. DBG简化:去掉无法继续连接和低覆盖度的分支,通常有如下几种情况:
1) 直接删除由于测序错误形成的低频K-mer;

2) 通过短序列将一些很短的重复解开,让每个节点的出入度都为1;

3) 如果Kmer1和Kmer2有很高的相似性,将形成的泡状结构合并;

D. 解图获得一致性序列:在简化图的基础上,仍然会因有很多分叉位点无法确定真正的连接关系,因此接下来的每个分叉位点将序列截断,得到contigs;
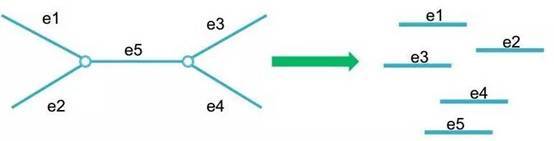
E. 构建scaffold: 将质控后的reads比对回上一步得到的congtigs,利用reads之间的连接关系和插入片段大小信息,将contigs连接成scaffolds;

F. Gap Close: 通过PE reads来填补scaffolds内部的Gap,经过Gap填补后,如果还有含N的Gap,则将该条scaffold在Gap处打断,并去掉N,形成最后的scaftigs;

3)OLC vs DBG
由于二代测序得到的reads长度较短,包含的信息量较少,因此完成基因组拼接需要较高的覆盖度。OLC算法适用于读长较长的序列组装,通过构成的OLC图寻找Consensus sequence的过程,实际上是哈密顿通路寻找的问题,算法非常复杂。
若采用OLC算法,会大大增加拼接的复杂性以及运算量。而采用DBG算法,通过K-1的overlap关系,构建DBG图,通过寻找欧拉路径得到Contig序列,从算法的角度极大的简化了组装的难度。
2. 组装软件
常用短reads(二代测序)组装软件比较:
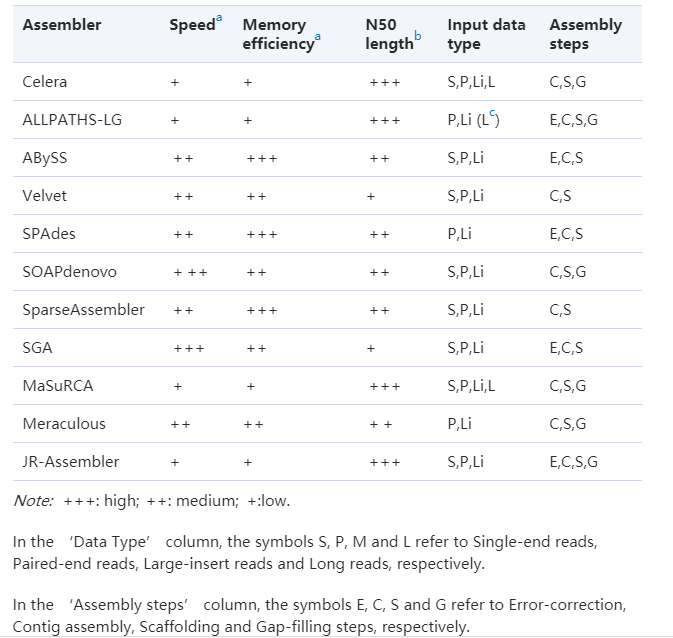
Jang-il Sohn, Jin-Wu Nam. The present and future of de novo whole-genome assembly
不同软件的组装结果差别可能很大,跟物种的基因组复杂度也有关系。所以有条件的话,最好选择几个软件,选择其中最好的结果,尤其是大基因组。最经典或使用频率最高的莫过于Allpaths-LG和SOAPdenovo。
常用长reads(三代测序)组装软件:
两款有代表性的软件:Canu和Falcon,基于OLC算法。在不同物种上各有优势。一般,简单基因组优先考虑Canu,复杂基因组优先考虑Falcon(与Falcon-Unzip实现无缝对接,适用杂合度较高或远亲繁殖或多倍体物种)。

Edward S. Rice and Richard E. Green. New Approaches for Genome Assembly and Scaffolding
陆续也有很多新的三代组装的软件开发出来,比如国产软件WTDBG(阮珏和李恒开发)以DBG算法为基础开发的模糊布鲁因图算法,运行速度快,内存占用小,对于重复序列含量高和杂合度高的物种可能出现错误的碱基合并。但该软件对数据量有限制,一般用于简单的基因组。
3. 组装策略
以往限于技术,采用的是一代或纯二代测序数据进行从头组装。基于二代测序的组装面临很多挑战:
- 短reads远远小于原来的分子长度(通过测更多样本,建库随机)
- 海量数据增加组装的计算复杂性(采用kmers算法,关键在于定义k)
- 测序错误导致组装错误,影响contig的长度(通过提高质控标准)
- 短reads难以区分基因组的重复序列(加大测序深度,双末端文库/大片段文库)
- 测序覆盖度不均一,影响统计检验和结果诊断(提高深度,保证随机)
三代测序对基因组组装具有天然优势,通过技术发展和成本降低,现在已经鲜有纯二代测序的组装了(另一个原因可能是常见的简单基因组物种已经差不多测了),一般是二三代数据结合,加上光学图谱,遗传图谱或者Hi-C等技术来进行基因组的组装,即PacBio+HiSeq +BioNano+10X+Hi-C 等多种平台完美搭配。目前各种测序技术对基因组组装的贡献:

如果要获得染色体级别组装的基因组,通常是先用Pacbio或Nanopore技术进行contig构建,然后利用10X genomics或Bionano技术将contig连接成scaffold(可选项,主要目的是纠错和把部分contig以gap的形式进行初步连接),最后利用Hi-C染色质构象捕获技术(准确度可以媲美早期的遗传图谱),将contig/scaffold连接成染色体级别。
如分别利用Illumina+10X Genomics+BioNano和Pacbio+BioNano两种策略对人的基因组进行组装,同时以Illumina+Fosmid-end组装策略作为对照。三者组装的指标见下表。

4. 组装项目实施
1)测序前的准备
搜集物种或已发表的近缘物种相关信息,比如基因组大小,基因组重复程度,GC含量和分布,杂合度等。
Survey分析,即将测序得到的 reads 打断成 K-mer,通过 K-mer 分析,从数学的角度评估基因组的大小,杂合以及重复等信息。并进行初步组装,从初步组装的 Contig 的 GC 分布图上,判断该物种是否有污染等信息,从而为后续组装策略的制定提供可靠的依据。
获取基因组大小
基因组大小的获取关系到对以后组装结果的大小的正确与否判断;基因组太大(>10Gb),超出了目前denovo组装基因组软件的对机器内存的要求,从客观条件上讲是无法实现组装的。查询植物基因组大小的网站:http://data.kew.org/cvalues,查询动物基因组大小的网站:http://www.genomesize.com/。
如果没有搜录,需要考虑通过流式细胞仪、基于福尔根染色、定量PCR、Kmer等方法来估计基因组大小。
杂合度估计
杂合度对基因组组装的影响主要体现在不能合并姊妹染色体,杂合度高的区域,会把两条姊妹染色单体都组装出来,从而造成组装的基因组偏大于实际的基因组大小。
一般是通过SSR在测序亲本的子代中检查SSR的多态性。杂合度如果高于0.5%,则认为组装有一定难度。杂合度高于1%则很难组装出来。杂和度估计一般通过kmer分析来做。降低杂合度可以通过很多代近交来实现。
杂合度高,并不是说组装不出来,而是说,装出来的序列不适用于后续的生物学分析。比如拷贝数、基因完整结构。
是否有遗传图谱可用
随着测序对质量要求越来越高和相关技术的逐渐成熟,遗传图谱也快成了denovo基因组的必须组成。构建遗传图构建相关概念可以参考这本书(The handbook of plant genome mapping: genetic and physical mapping )
生物学问题的调研
实验设计很关键。
2) 测序样品准备
确定第一步没问题,就意味着这个物种是可以尝试测序的。测序样品对一些物种也是很大问题的,某些物种取样本身就是一个挑战的问题。
基因组测序用的样品最好是来自于同一个个体,这样可以降低个体间的杂合对组装的影响。大片段对此无要求。原则上进行 Survey 和de novo使用的 DNA 是来自一个个体的。如果DNA量不足以满足整个de novo项目,则建议小片段文库的DNA必须来自同一个体,三代大片段甚至超长片段的DNA文库使用同一群体的另一个个体。
3)测序策略的选择
根据基因组大小和具体情况选择个大概的k值,确定用于构建contig所需的数据量以及文库数量。对于植物基因组一般考虑的是大kmer(>31),动物的话一般在27左右,具体根据基因组情况调整。需要在短片段数据量达到20X左右的时候进行kmer分析。Kmer分析正常后,继续加测数据以达到最后期望的数据量。
文库构建,一般都是用不同梯度的插入片段来测序,小片段(200,500,800)和大片段(1k, 2kb 5kb 10kb 20kb 40kb)。如果是杂合度高和重复序列较多的物种,可能要采取fosmid-by-fosmid或者fosmid pooling的策略。
采用更多不同的技术组合见3.组装策略部分。
4)质控、基因组组装、质量评估
- 组装流程:原始数据-数据过滤-纠错-kmer分析-denovo组装
- 质控常用软件:FastQC,fastp,BFC,SOAPnuke(适合中小项目),SOAPfilter(适合大基因组)等。
- 纠错软件:COPE(An accurate k-mer based pair-end reads connection tool to facilitate genome assembly,doi: 10.1093/bioinformatics/bts563)
- kmer分析软件:jellyfish,kmerfreq,kmerscan等。
- 主流组装软件:ALLPATHS-LG,SOAPdenovo2,ABySS,Velvet,Minia,MasuRCA,Rabbit(overlap组装)。组装并非一次就能得到理想的结果,会根据已有的组装结果做分析,调整参数,处理数据,加测少量数据等策略来得到比较理想的结果。
- 补洞软件:GapCloser,KGF等。
- 组装评价软件:BUSCO,Quast等。包括序列一致性(比对和覆盖度)、序列完整性(EST数据或RNA)、准确性(全长BAC序列和scaffold是否具好的一致性)、保守性基因(保守蛋白家族集合)等方面进行评估。
5)基因组注释
一般包括重复序列,基因结构,基因功能,非编码RNA注释。后续再专门总结。
6)生物学分析
主要是比较基因组分析,一般包括基因家族,系统进化,正选择,共线性等分析。还有针对物种自身特点的个性化分析。后续专门总结。
7)更多参考内容
基因组组装 by Life Intelligence
5. 动植物Denovo测序项目的主要分析内容
基因组Survey:
- K-mer分析以及基因组大小估算
- 杂合率估算
- 初步组装
- GC-Depth分布分析
基因组组装:
- 组装
- GC-Depth分布分析
- GC含量分布分析
- 测序深度分析
- 常染色体区域覆盖度评估(需提供BAC或Fosmid序列)
- 基因区覆盖度评估(需提供EST或转录组序列)
基因组注释:
- repeat注释
- 基因预测
- 基因功能注释
- ncRNA 注释
进化分析:
- 基因聚类分析(也叫基因家族鉴定,动物TreeFam;植物OrthoMCL)
- 物种系统发育树构建
- 物种分歧时间估算(需要标定时间信息)
- 基因组共线性分析
- 全基因组复制分析(动物WGAC;植物WGD)
参考资料:
从零开始生物信息学(5):基因组组装
六步教会你基因组组装!
干货:最全面的三代基因组之组装篇(上)
纯二代测序从头组装基因组
三代组装软件简介
生信老司机教你如何做基因组项目
基因组测序、组装与分析总结
动植物 de novo 测序产品服务(华大)
全基因组de novo测序产品服务(诺禾)
二代测序组装PK三代测序组装
基因组组装结果质量评估