scATAC分析神器ArchR初探-简介(1)
scATAC分析神器ArchR初探-ArchR进行doublet处理(2)
scATAC分析神器ArchR初探-创建ArchRProject(3)
scATAC分析神器ArchR初探-使用ArchR降维(4)
scATAC分析神器ArchR初探--使用ArchR进行聚类(5)
scATAC分析神器ArchR初探-单细胞嵌入(6)
scATAC分析神器ArchR初探-使用ArchR计算基因活性值和标记基因(7)
scATAC分析神器ArchR初探-scRNA-seq确定细胞类型(8)
scATAC分析神器ArchR初探-ArchR中的伪批次重复处理(9)
scATAC分析神器ArchR初探-使用ArchR-peak-calling(10)
scATAC分析神器ArchR初探-使用ArchR识别标记峰(11)
scATAC分析神器ArchR初探-使用ArchR进行主题和功能丰富(12)
scATAC分析神器ArchR初探-利用ArchR丰富ChromVAR偏差(13)
scATAC分析神器ArchR初探-使用ArchR进行足迹(14)
scATAC分析神器ArchR初探-使用ArchR进行整合分析(15)
scATAC分析神器ArchR初探-使用ArchR进行轨迹分析(16)
15-使用ArchR进行整合分析
ArchR的主要优势之一是它能够集成多个级别的信息以提供新颖的见解。这可以采取仅ATAC-seq的分析形式,例如识别峰的可共同接近性以预测调节相互作用,或采用整合scRNA-seq数据的分析,例如通过峰-基因连锁分析预测增强子活性。无论哪种情况,ArchR都可以轻松地从scATAC-seq数据中获得更深刻的见解。
15.1创建单元格的低重叠聚合
ArchR促进了许多涉及特征相关性的综合分析。在稀疏的单细胞数据中执行这些计算会导致这些相关分析中的大量噪声。为了克服这一挑战,我们采用了Cicero引入的方法在这些分析之前创建单个细胞的低重叠聚集体。为了减少偏差,我们会过滤与其他任何聚合具有大于80%重叠的聚合。为了提高这种方法的速度,我们使用“ Rcpp”包开发了一种优化的迭代重叠检查例程的实现,以及C ++中快速特征关联的实现。这些优化的方法用于ArchR中,用于计算峰的可及性,峰与基因的链接以及其他链接分析。这些低重叠的聚合的使用是在引擎盖下进行的,但是为了清楚起见,我们在此提及。
15.2与ArchR的可共访问性
共可及性是许多单个细胞中两个峰之间可及性的关联。换句话说,当在单个单元中可以访问峰A时,通常也可以访问峰B。我们在下面以视觉方式说明了这一概念,表明Enhancer E3通常与Promoter P可以同时访问。
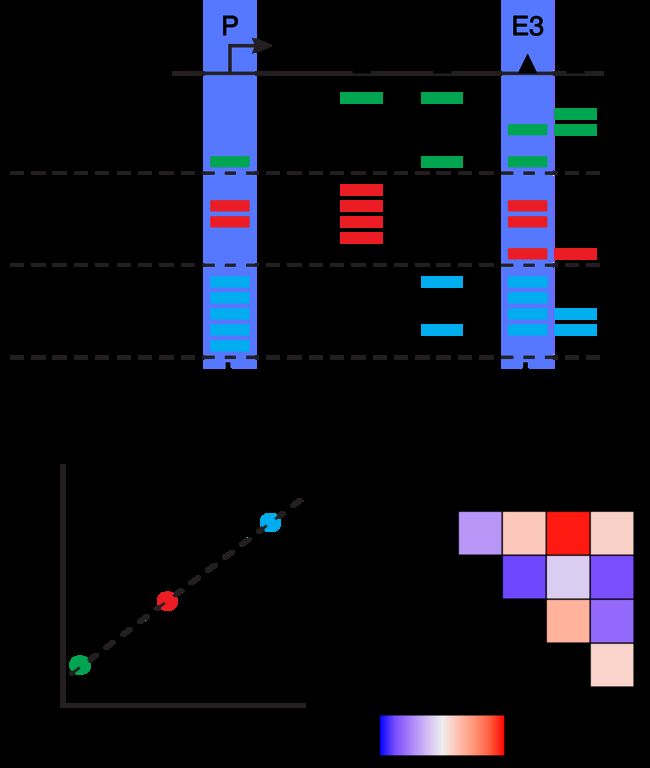
关于协同访问分析,要注意的一件事是,它经常将特定于细胞类型的峰鉴定为协同访问。这是因为这些峰通常在单个单元格类型中都可以一起访问,而在所有其他单元格类型中通常都不能访问。这会产生很强的相关性,但不一定意味着这些峰之间存在调节关系。
要计算ArchR中的可访问性,我们使用addCoAccessibility()将峰值可访问性信息存储在中的功能ArchRProject。
projHeme5 <- addCoAccessibility(
ArchRProj = projHeme5,
reducedDims = "IterativeLSI"
)
我们可以ArchRProject通过getCoAccessibility()函数从中检索此可访问性信息,该函数返回DataFrame对象if returnLoops = FALSE。
cA <- getCoAccessibility(
ArchRProj = projHeme5,
corCutOff = 0.5,
resolution = 1,
returnLoops = FALSE
)
其中DataFrame包含一些重要的信息。的queryHits和subjectHits列表示被发现相关联,这两个峰的索引。该correlation列给出了这两个峰之间可及性的数字相关性。
cA
这种可共访问性DataFrame还具有包含GRanges相关峰对象的元数据组件。上面提到的queryHits和的索引subjectHits适用于此GRanges对象。
metadata(cA)[[1]]
如果我们设置returnLoops = TRUE,则将getCoAccessibility()以循环跟踪的形式返回可访问性信息。在此GRanges对象中,IRanges每次互动的图谱的起点和终点分别指向两个不同的可同时访问的峰。该resolution参数设置这些循环的碱基对分辨率。当为时resolution = 1,这将创建连接每个峰中心的循环。
cA <- getCoAccessibility(
ArchRProj = projHeme5,
corCutOff = 0.5,
resolution = 1,
returnLoops = TRUE
)
我们可以将此GRanges对象与DataFrame上面生成的对象进行比较。
cA[[1]]
相反,如果我们降低到的循环的分辨率resolution = 1000,则可能有助于过度绘制协同访问交互。在下面,我们看到GRanges对象中的总条目比上面的少。
cA <- getCoAccessibility(
ArchRProj = projHeme5,
corCutOff = 0.5,
resolution = 1000,
returnLoops = TRUE
)
cA[[1]]
同样,如果我们进一步降低分辨率resolution = 10000,我们将确定更少的协同访问交互。
cA <- getCoAccessibility(
ArchRProj = projHeme5,
corCutOff = 0.5,
resolution = 10000,
returnLoops = TRUE
)
cA[[1]]
15.2.1绘制浏览器的可共通性轨迹
在向我们添加了可访问性信息后,我们ArchRProject就可以在绘制浏览器轨迹时将其用作循环轨迹。我们通过函数的loops参数来实现plotBrowserTrack()。在这里,我们使用的是默认参数getCoAccessibility(),其中包括corCutOff = 0.5,resolution = 1000,和returnLoops = TRUE。
markerGenes <- c(
"CD34", #Early Progenitor
"GATA1", #Erythroid
"PAX5", "MS4A1", #B-Cell Trajectory
"CD14", #Monocytes
"CD3D", "CD8A", "TBX21", "IL7R" #TCells
)
p <- plotBrowserTrack(
ArchRProj = projHeme5,
groupBy = "Clusters2",
geneSymbol = markerGenes,
upstream = 50000,
downstream = 50000,
loops = getCoAccessibility(projHeme5)
)
为了绘制浏览器轨迹,我们使用该grid.draw函数,并使用$访问器按名称选择特定的标记基因。
grid::grid.newpage()
grid::grid.draw(p$CD14)
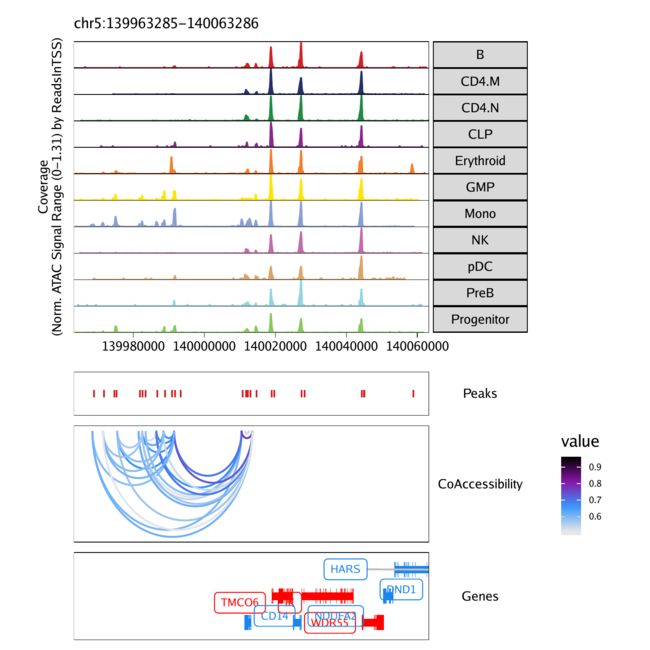
要保存此图的可编辑矢量化版本,请使用plotPDF()。
plotPDF(plotList = p,
name = "Plot-Tracks-Marker-Genes-with-CoAccessibility.pdf",
ArchRProj = projHeme5,
addDOC = FALSE, width = 5, height = 5)
15.3 Peak2Gene与ArchR的链接
与协同访问相似,ArchR也可以识别所谓的“峰到基因链接”。峰与基因之间的链接与共同访问之间的主要区别在于,共同访问是仅ATAC-seq的分析,用于寻找两个峰之间的可访问性的相关性,而峰到基因的链接则利用整合的scRNA-seq数据寻找峰可及性与基因表达之间的相关性。这些代表解决类似问题的正交方法。但是,由于峰与基因之间的联系将scATAC-seq和scRNA-seq数据相关联,因此我们经常认为这些联系与基因调控相互作用更为相关。
为了识别ArchR中的峰到基因链接,我们使用该addPeak2GeneLinks()函数。
projHeme5 <- addPeak2GeneLinks(
ArchRProj = projHeme5,
reducedDims = "IterativeLSI"
)
然后,我们可以使用类似于使用getPeak2GeneLinks()函数检索共通性交互的方式来检索这些峰-基因链接。如我们先前所见,此功能允许用户指定链接的关联和分辨率的截止值。
p2g <- getPeak2GeneLinks(
ArchRProj = projHeme5,
corCutOff = 0.45,
resolution = 1,
returnLoops = FALSE
)
当returnLoops设置为false时,此函数将返回与所返回的DataFrame对象类似的DataFrame对象getCoAccessibility()。主要区别在于,scATAC-seq峰idxATAC的索引存储在列中,而scRNA-seq基因的索引存储在idxRNA列中。
p2g
这种峰到基因的链接DataFrame还具有包含GRanges相关峰对象的元数据组件。idxATAC上面提到的索引适用于此GRanges对象。
metadata(p2g)[[1]]
如果设置returnLoops = TRUE,getPeak2GeneLinks()则将返回GRanges连接峰和基因的循环跟踪对象。至于共通性,IRanges对象的开始和结束代表峰的位置和被链接的基因。当为时resolution = 1,这会将峰的中心链接到基因的单碱基TSS。
p2g <- getPeak2GeneLinks(
ArchRProj = projHeme5,
corCutOff = 0.45,
resolution = 1,
returnLoops = TRUE
)
p2g[[1]]
我们可以通过设置来降低这些链接的分辨率resolution = 1000。这在将链接绘制为浏览器轨迹时非常有用,因为在某些情况下,许多附近的峰都链接到同一基因,这可能很难可视化。
p2g <- getPeak2GeneLinks(
ArchRProj = projHeme5,
corCutOff = 0.45,
resolution = 1000,
returnLoops = TRUE
)
p2g[[1]]
降低分辨率甚至会进一步减少所识别的峰到基因链接的总数。
p2g <- getPeak2GeneLinks(
ArchRProj = projHeme5,
corCutOff = 0.45,
resolution = 10000,
returnLoops = TRUE
)
p2g[[1]]
15.3.1使用峰到基因链接绘制浏览器轨迹
要将这些峰到基因链接绘制为浏览器轨迹,我们使用上一部分中所示的可共访问性相同的工作流程。在这里我们使用plotBrowserTrack()功能
markerGenes <- c(
"CD34", #Early Progenitor
"GATA1", #Erythroid
"PAX5", "MS4A1", #B-Cell Trajectory
"CD14", #Monocytes
"CD3D", "CD8A", "TBX21", "IL7R" #TCells
)
p <- plotBrowserTrack(
ArchRProj = projHeme5,
groupBy = "Clusters2",
geneSymbol = markerGenes,
upstream = 50000,
downstream = 50000,
loops = getPeak2GeneLinks(projHeme5)
)
为了绘制浏览器轨迹,我们使用该grid.draw函数,并使用$访问器按名称选择特定的标记基因。
grid::grid.newpage()
grid::grid.draw(p$CD14)
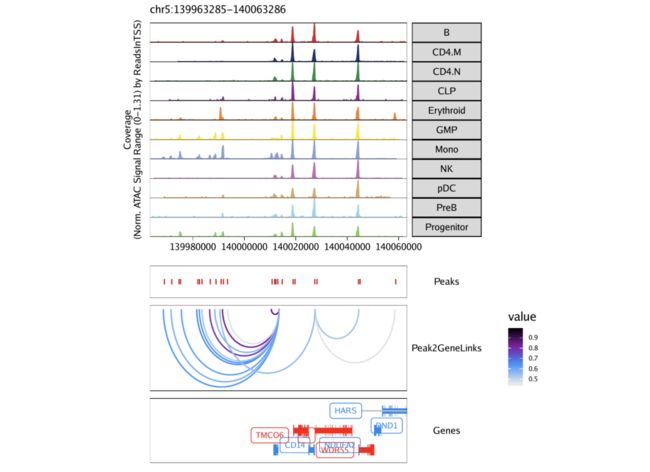
To save an editable vectorized version of this plot, we use plotPDF().
plotPDF(plotList = p,
name = "Plot-Tracks-Marker-Genes-with-Peak2GeneLinks.pdf",
ArchRProj = projHeme5,
addDOC = FALSE, width = 5, height = 5)
15.3.2 Plotting a heatmap of peak-to-gene links
To visualize the correspondence of all of our peak-to-gene links, we can plot a peak-to-gene heatmap which contains two side-by-side heatmaps, one for our scATAC-seq data and one for our scRNA-seq data. To do this, we use the plotPeak2GeneHeatmap()
p <- plotPeak2GeneHeatmap(ArchRProj = projHeme5, groupBy = "Clusters2")
The heatmap rows are clustered using k-means clustering based on the value passed to the parameter k, which defaults to 25 as shown below.
p
15.4确定正TF调节器
ATAC-seq可以无偏鉴定TF,这些TF在包含其DNA结合基序的位点上,染色质可及性表现出很大的变化。但是,当通过位置权重矩阵(PWM)进行汇总时,TF家族(例如GATA因子)在其绑定图案中具有相似的功能。

这种基序相似性使其难以鉴定可能导致观察到的染色质可及性在其预测的结合位点发生变化的特定TF。为了规避这一挑战,我们先前使用了ATAC-seq和RNA-seq来鉴定其基因表达与其相应基序可及性变化呈正相关的TF。我们将这些TF称为“积极监管者”。但是,该分析依赖于并非在所有实验中都容易获得的匹配基因表达数据。为了克服这种依赖性,ArchR可以识别其推断的基因得分与其chromVAR TF偏差z得分相关的TF。为此,ArchR将TF基序的chromVAR偏差z分数与来自低重叠细胞聚集体的TF基因的基因活性得分相关联。将scRNA-seq与ArchR结合使用时,
15.4.1第1步。确定偏差TF图案
鉴定阳性TF调节剂的第一部分是鉴定异常TF基序。我们在上一章中进行了此分析,MotifMatrix为所有图案创建了一个chromVAR偏差和z分数偏差。我们可以使用getGroupSE()返回a 的函数来获得按聚类平均的数据SummarizedExperiment。
seGroupMotif <- getGroupSE(ArchRProj = projHeme5, useMatrix = "MotifMatrix", groupBy = "Clusters2")
由于此SummarizedExperiment对象来自,因此MotifMatrix具有两个序列名称-“偏差”和“ z”-对应于chromVAR的原始偏差和z分数偏差。
seGroupMotif
我们可以将其子集化为SummarizedExperiment偏差z得分。
seZ <- seGroupMotif[rowData(seGroupMotif)$seqnames=="z",]
然后,我们可以确定所有群集之间z得分的最大增量。这将有助于根据跨群集观察到的变化程度对主题进行分层。
rowData(seZ)$maxDelta <- lapply(seq_len(ncol(seZ)), function(x){
rowMaxs(assay(seZ) - assay(seZ)[,x])
}) %>% Reduce("cbind", .) %>% rowMaxs
15.4.2步骤2.识别相关的TF基序和TF基因得分/表达
为了鉴定其基序可及性与其自身基因活性相关的TF(通过基因评分或基因表达),我们使用该correlateMatrices()功能并提供我们感兴趣的两个矩阵,在这种情况下为GeneScoreMatrix和MotifMatrix。如前所述,这些相关性是在reducedDims参数中指定的较低维空间中确定的许多低重叠单元聚合中确定的。
corGSM_MM <- correlateMatrices(
ArchRProj = projHeme5,
useMatrix1 = "GeneScoreMatrix",
useMatrix2 = "MotifMatrix",
reducedDims = "IterativeLSI"
)
此函数返回一个DataFrame对象,其中包含来自每个矩阵的元素以及低重叠单元格聚合之间的相关性。
corGSM_MM
我们可以使用GeneIntegrationMatrix代替进行相同的分析GeneScoreMatrix。
corGIM_MM <- correlateMatrices(
ArchRProj = projHeme5,
useMatrix1 = "GeneIntegrationMatrix",
useMatrix2 = "MotifMatrix",
reducedDims = "IterativeLSI"
)
corGIM_MM
步骤3.将最大Delta偏差添加到相关数据帧
对于这些相关分析中的每一个,我们都可以使用在步骤1中计算出的聚类之间观察到的最大增量来注释每个基序。
corGSM_MM$maxDelta <- rowData(seZ)[match(corGSM_MM$MotifMatrix_name, rowData(seZ)$name), "maxDelta"]
corGIM_MM$maxDelta <- rowData(seZ)[match(corGIM_MM$MotifMatrix_name, rowData(seZ)$name), "maxDelta"]
步骤4.确定正TF调节器
我们可以使用所有这些信息来确定正TF调节器。在以下示例中,我们将正调节剂视为其基序与基因得分(或基因表达)之间的相关性大于0.5,且调整后的p值小于0.01,且z值偏差的最大簇间差异为正的那些TF在前四分之一。
我们应用这些选择标准,并进行一些文字处理以隔离TF名称。
corGSM_MM <- corGSM_MM[order(abs(corGSM_MM$cor), decreasing = TRUE), ]
corGSM_MM <- corGSM_MM[which(!duplicated(gsub("\\-.*","",corGSM_MM[,"MotifMatrix_name"]))), ]
corGSM_MM$TFRegulator <- "NO"
corGSM_MM$TFRegulator[which(corGSM_MM$cor > 0.5 & corGSM_MM$padj < 0.01 & corGSM_MM$maxDelta > quantile(corGSM_MM$maxDelta, 0.75))] <- "YES"
sort(corGSM_MM[corGSM_MM$TFRegulator=="YES",1])
从基因得分和基序偏差z分数中识别出这些阳性TF调节剂后,我们可以在点图中突出显示它们。
p <- ggplot(data.frame(corGSM_MM), aes(cor, maxDelta, color = TFRegulator)) +
geom_point() +
theme_ArchR() +
geom_vline(xintercept = 0, lty = "dashed") +
scale_color_manual(values = c("NO"="darkgrey", "YES"="firebrick3")) +
xlab("Correlation To Gene Score") +
ylab("Max TF Motif Delta") +
scale_y_continuous(
expand = c(0,0),
limits = c(0, max(corGSM_MM$maxDelta)*1.05)
)
p
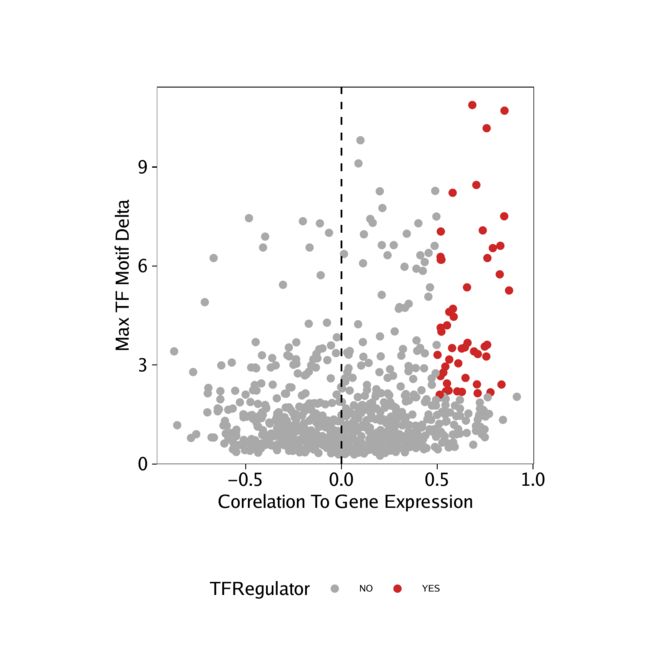
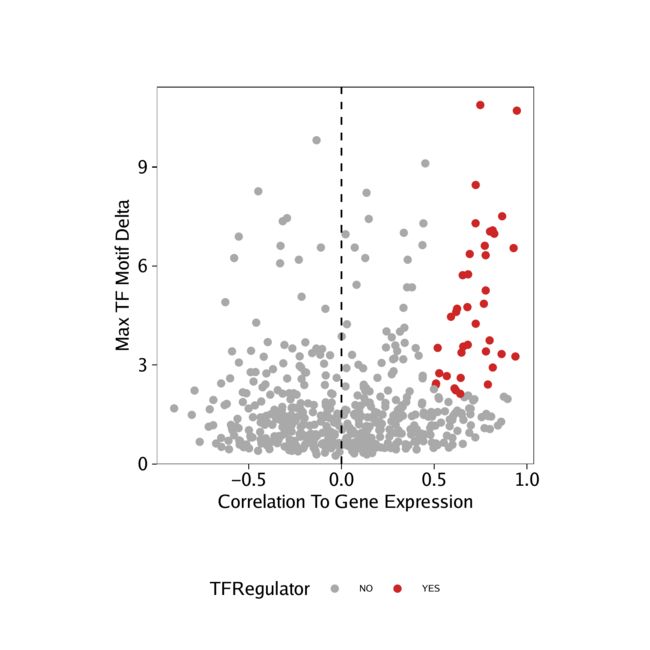
我们可以对从得出的相关性执行相同的分析GeneIntegrationMatrix。
corGIM_MM <- corGIM_MM[order(abs(corGIM_MM$cor), decreasing = TRUE), ]
corGIM_MM <- corGIM_MM[which(!duplicated(gsub("\\-.*","",corGIM_MM[,"MotifMatrix_name"]))), ]
corGIM_MM$TFRegulator <- "NO"
corGIM_MM$TFRegulator[which(corGIM_MM$cor > 0.5 & corGIM_MM$padj < 0.01 & corGIM_MM$maxDelta > quantile(corGIM_MM$maxDelta, 0.75))] <- "YES"
sort(corGIM_MM[corGIM_MM$TFRegulator=="YES",1])
p <- ggplot(data.frame(corGIM_MM), aes(cor, maxDelta, color = TFRegulator)) +
geom_point() +
theme_ArchR() +
geom_vline(xintercept = 0, lty = "dashed") +
scale_color_manual(values = c("NO"="darkgrey", "YES"="firebrick3")) +
xlab("Correlation To Gene Expression") +
ylab("Max TF Motif Delta") +
scale_y_continuous(
expand = c(0,0),
limits = c(0, max(corGIM_MM$maxDelta)*1.05)
)
p
参考材料:
https://www.archrproject.com/