内皮活性增强子的重构与肺动脉高压中异常基因调控网络相关
Remodeling of active endothelial enhancers is associated with aberrant gene-regulatory networks in pulmonary arterial hypertension
https://www.nature.com/articles/s41467-020-15463-x
•研究背景
•实验方法
•实验结果
•结论与讨论
什么是肺动脉高压?
目前报道约15%肺动脉高压(PAH)患者为家族性。其中70%的人携带BMPR2基因突变。另外30%的散发性PAH病例也有BMPR2突变,统称为遗传性(H)PAH。还有一类不知道原因的,统称为特发性(I)PAH。
患者症状表现为疲倦和呼吸急促,有时会持续数年,但由于这些患者症状不够典型(很多疾病都有此表现),在疾病进入病理晚期之前,PAH的诊断往往比较困难。
目前的治疗,包括皮下或静脉注射前列环素,本质上都是扩张血管的;虽然它们大大降低了死亡率,改善了生活质量,但PAH的5年存活率低于60%。
Disease: pulmonary arterial hypertension (PAH)
-
Object: pulmonary arterial endothelial cells (PAECs)
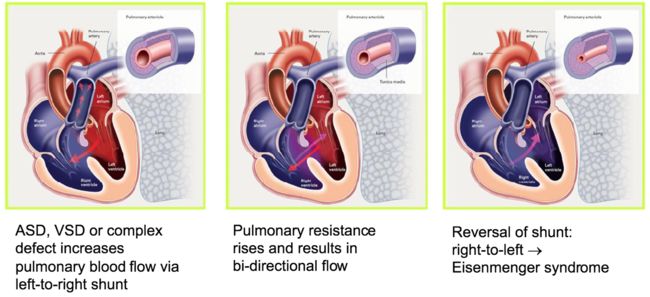
下面是肺动脉高压进展的模式图,由于近端动脉的闭塞和远端微血管的丢失,肺循环中的压力逐渐升高,最终导致右侧心力衰竭。
怎么进行研究
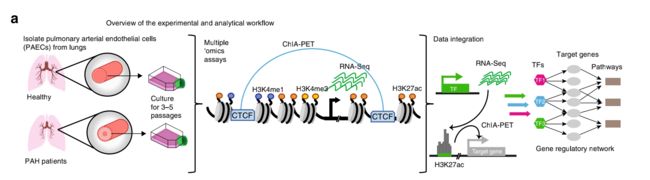
大多数特发性PAH患者没有已知的原因突变,因此,作者试图通过研究PAH患者与健康对照组疾病相关组织中染色质标记和基因表达的分子差异来评估潜在的非编码基因调控元件的变异对疾病机制的影响。
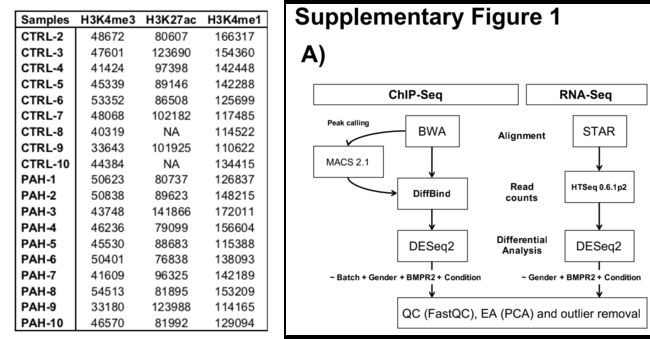
对来自遗传性肺动脉高压(HPAH;携带BMPR2突变;n=2)和特发性肺动脉高压(IPAH;未知突变;n=8)和对照组(n=9;图1A,补充图1A)共19例来源的小肺动脉内皮细胞(PAECs)进行三个活性组蛋白标记(H3K27ac,H3K4me1,H3K4me3)和RNA-Seq进行了芯片测序序列分析。
从患者肺移植过程中采集的PAECs细胞培养3-5代,以获得CHIP-SEQ实验所需的数量,并尽量降低批次效应。
补充一下背景,参考https://www.jianshu.com/p/f7d7bf8aec0b
使用到的样本信息及分析软件:
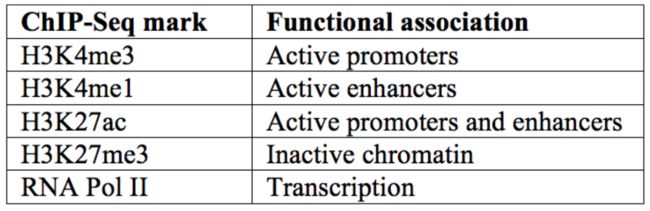
使用配对末端标签测序的染色质相互作用分析技术CHIA-PET (Chromatin interaction analysis using paired-end tag sequencing. )
配对末端标签测序分析染色质相互作用(chromatin interaction analysis by paired-end tag sequencing,ChIA-PET)技术是一项在全基因组范围内分析远程染色质相互作用的新技术。它把染色质免疫沉淀(chromatin immunoprecipitation,ChIP)技术、染色质邻近式连接(chromatin proximity ligation)技术、配对末端标签(paired-endtag,PET)技术和新一代测序(next-generation sequencing)技术融为一体,在基因组三维折叠和loop状态下分析基因表达和调控。
染色质远程交互在转录调控中发挥着重要作用。2002年起,染色质构象捕获技术(3C)及其衍生方法不断发展,该方法用于揭示DNA与DNA之间的空间位置关系。基于配对末端标签测序的染色质交互分析技术(ChIA-PET)将3C与ChIP相结合,可用于描绘全基因范围内特定蛋白介导的染色质交互图谱。ChIA-PET Tool为一个ChIA-PET数据处理的软件,最初于2010年公开发表。
下面这张图是搜索自网络介绍CHIA-PET背景相关示意图,及相关数据分析软件的。

下面正式开始正文的描述:
研究流程如下:
从患者和病人肺动脉分离PAECs进行培养3-5代后进行CTCF的CHIA-PET实验与活性增强子调控的靶基因进行疾病相关基因调控网络分析。
研究结果如下:
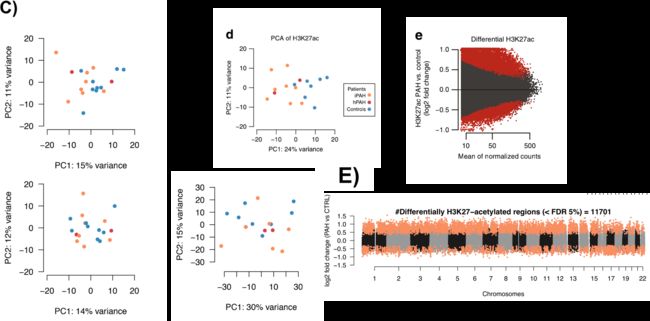
在PAH患者中发现活性增强子(H3K27ac)的广泛重构。而通用增强子标记(H3K4me1)、启动子标记(H3K4me3)和基因表达(RNAseq)没有明显变化。这表明发生改变的是活性增强子。
1.Extensive remodeling of H3K27ac in PAH patients vs controls
染色质标记pattern符合预期:H3K4me3和H3K27ac的peaks在转录起始点(TSS)富集,并与基因表达水平正相关(Pearson‘s R=0.51和R=0.59),而H3K4me1的peaks更远离TSS且与RNA相关性较低(R=0.22)。作者观察到HPAH患者与IPAH之间相似的Fold change变化(补充图1F),这表明分子检测之间的信号是大致一致的(图1b,c)。因此作者将IPAH和HPAH患者结合起来进行所有后续分析。

对每种组蛋白标记和RNAseq的差异区域(前1000个变异最大的peaks/基因)进行的主成分分析(PCA)表明,只有H3K27ac能够将样本分为PAH患者和健康对照组(图1d;补充图1C)。
作者还发现,在IPAH和对照(图1e和补充图1E)之间,有数千个位点在H3K27ac有显著的差异变化(FDR<0.05)的11,701个),而在H3K4me1和H3K4me3的差异变化的peaks很少(在FDR<5%处没有发现差异表达基因,在<10%处只有少数区域显著(图未展示))。
由于前人的研究已经在不同的样本数据集中[9-11]已经确定了一些在PAH中的差异表达基因,因此作者在部分样品(两个IPAH,两个HPAH,四个对照)中对一些基因(图1f)的基线表达水平进行了qPCR验证,这些基因被选择的原因是其启动子中具有一系列差异H3K27ac信号(图1g)。
在qPCR检测中,它们在基线基因表达水平上都没有显示出任何差异。但当作者比较组蛋白标记和RNAseq的样品之间的成对相关性分析时发现RNAseq的变异水平要高得多,无论这些样本来自PAH还是对照,这表明缺乏差异表达的基因可能部分是由样本的异质性导致的(补充图1G)。
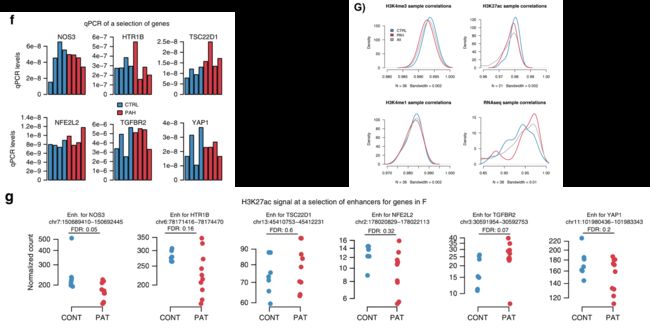
为了获得对差异修饰的H3K27ac peaks的功能检测,我们使用表观基因组项目(Epigenomic roadmap project)[12]的数据评估了它们的染色质状态。
我们对人脐静脉内皮细胞(HUVEC)使用了chromHMM状态分析,HUVEC是与肺动脉内皮细胞最相似的细胞系。正如对活性标记的预测,发现在测序结果中H3K27ac的peaks主要落在活性调控区域(包括启动子和增强子),而很少在异染色质中(图1h,补充图1H)。
此外,PAH患者PAECs中差异H3K27ac的peaks仅在活性增强子中富集(adjusted p-value (adj. p-val) < 1E − 16; Odds Ratio (OR) = 4.4),甚至在活性启动子中轻度缺失(adj. p-val = 0.03; OR = 0.93; 图1g)。
综上所述,这些结果表明,尽管PAH患者和健康对照组的PAEC在基因表达方面相似,但它们拥有活性染色质区域的大规模重塑,特别是在活性增强子上。这可能表明,当PAH患者PAECs仍处于内皮状态时,PAECs可能已经准备好对外界刺激做出不同于健康细胞的反应。
2.Differentially H3K27-acetylated regions are disease relevant.
为了描述差异修饰区域和与PAH相关的分子机制,作者对可能受差异H3K27乙酰化区域调控的基因进行了功能富集分析。
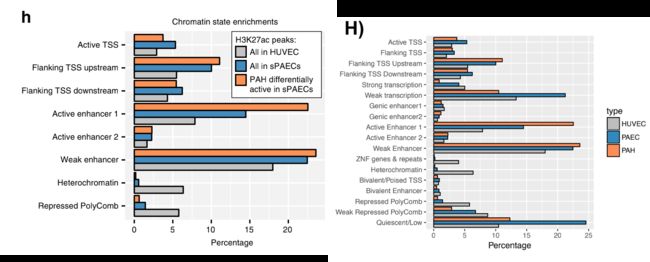
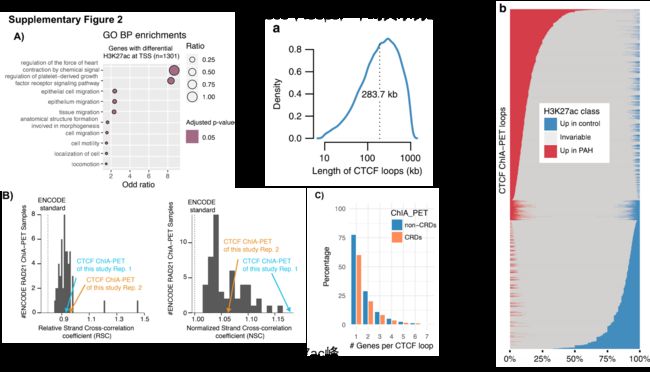
然而,与启动子中差异修饰区域的轻度缺失一致,作者发现启动子标记的差异peaks调控的基因没有被富集到特定GO通路,只有少数非常广泛的H3K27-ac标记启动子peaks调控的基因富集到了几条内皮增殖分化相关的通路(补充图2A)。
因此,由于差异的H3K27ac peaks富集在增强子,我们必须设计一种策略,将H3K27ac调节元件与其靶标基因联系起来。
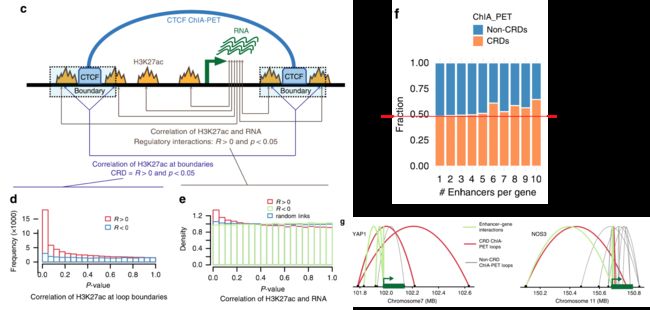
随后作者用CTCF作为锚定因子,通过成对末端标签测序(CHIA-PET)对来自健康供者的肺动脉内皮细胞中的染色质相互作用进行了分析。得到的数据质量高于ENCODE标准,与ENCODE CHIA-PET(补充图2B)的范围相似。共产生了21,805个loops,平均大小为283.7 kb(图2a),每个loop平均包含2个基因和25个H3K27ac峰(补充图2C)。
作者观察到CHIA-PET分析得到的loop中的H3K27ac peaks表现为一种高度协调的方式:在患者和对照组之间要么全部上调,要么全部下调(图2b)。这种与PAH相关的染色质变化在整个CTCF loop上的协同作用表明,它们可能作为独立的调控元件在PAH发生发展中发挥作用。因此,也说明不需要增强子-启动子的直接接触,而是寻找可以使其高度协调的loops,或称之为染色质调节结构域(CRD),可作为搜索增强子-靶基因相互作用的基础。那么怎么去识别这种loops(CRD)呢?使用CHIA-PET loops两个边界上的H3K27ac信号的相关性作为该loops的协调强度的替代指标,假设如果一个loop的最远位置的H3K27ac peak是相关的,那么中间的所有peak也将是相关的。
Define of CRD
当边界H3K27ac峰与p值<0.05呈正相关时,loops被定义为CRD(图2C)。最近在淋巴母细胞系[13]中也提出了类似的CRDs概念。
作为统计验证,结果显示负相关的p值分布没有信号,而正相关在低p值时被富集(图2d)。
然后,为了将基因与其特定的调控元件(增强子、启动子)联系起来,作者利用每个CRD中每个基因-peaks对的H3K27ac信号与RNA表达之间的Pearson相关性,并将正相关(p值为0.05)定义为调控、增强子-基因和启动子-基因的相互作用,假定增强子和基因表达应该是正相关的。
作者发现了更多的正相关信号,而负相关和随机遗传peaks链接的p值分布显示没有信号(图2e)。
每个基因的增强子数量,被定义为与基因表达相关的H3K27ac峰,在CRD中比非CRD 中loops比例高,从而验证了我们的假设(图2f,补充图2C,d)。
CRD内的基因示例如图2g和补充图2E所示。
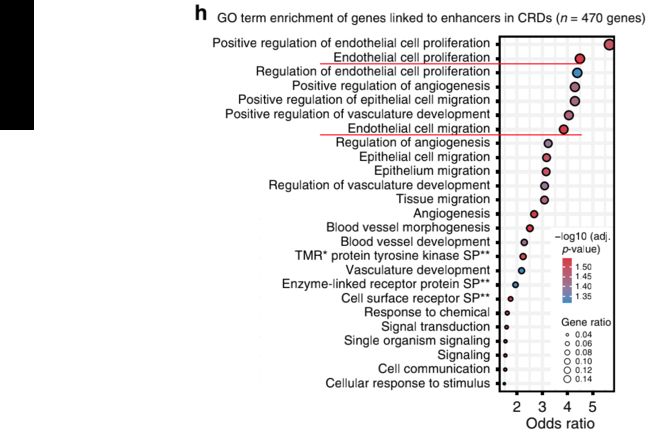
为了评估CRD的生物学意义,我们对CRD中与不同活性元件相关的基因进行了GO富集分析,并将其与非CRD 中loop相关的基因的GO分析进行了比较。
对于与CRD相关的基因,我们发现与PAH相关的内皮生物学和过程有很强的富集性,如“内皮细胞增殖”(adj. p-value = 0.03, OR = 4.5)和“内皮细胞迁移”(adj. p-value = 0.03, OR = 3.8;图2H),而对于非CRD连锁基因,没有GO terms显著富集。
这突出了CRD对于研究增强子-基因调控相互作用的生物学相关性。
总体而言,这些结果表明,通过CRD将基因与PAH中不同活性的增强子联系起来,捕获了预期的疾病相关和细胞类型特异性信号,并说明了将增强子与其目标基因联系起来对理解疾病机制的重要性。
同时,发现这些内皮细胞和PAH特异的功能,证明了我们的数据和分析方法的适用性,可以更多地了解PAH涉及的病理生物学。
4.Known PAH TFs are differentially active in PAH vs controls.
考虑到CRD内内皮功能和疾病特异性GO通路的不同活性区域的强烈富集,作者想更深入挖掘这些富集的分子通路的相关机制。
根据之前一项研究的结果,作者发现调控组蛋白修饰水平的遗传变异通常位于TF结合位点内[14],因此作者提出一个假设:H3K27ac信号的差异是由一组特定的与疾病相关的TF驱动的。并且他们最近开发一个软件diffTF的工具,并正式提出了这一想法,该工具基于两组样本之间的全基因组H3K27ac信号来估计差异TF活性[8]。
简而言之,diffTF将H3K27ac信号的差异聚集在给定TF的所有假定结合位点附近,作为其差异活性的估计(见方法,补充图3A,B)。在这里,我们应用diffTF来量化PAH患者和对照组之间在我们数据中PAECs表达的所有TF的差异活动。
总共,我们发现了318个差异活性因子(代表了640个测试基序中的339个;FDR 0.001;图3a,补充数据3)。
有趣的是,当作者将TF分为激活因子和抑制因子时(在分类模式[8]中使用diffTF),他们发现患者和对照之间的差异TF活性和差异表达之间总体上存在显著的相关性(补充图3B)。
这表明TF表达的微小变化可以导致可检测到的活性差异,这可能是由于H3K27ac信号在所有结合位点上的聚集所导致的[8]。
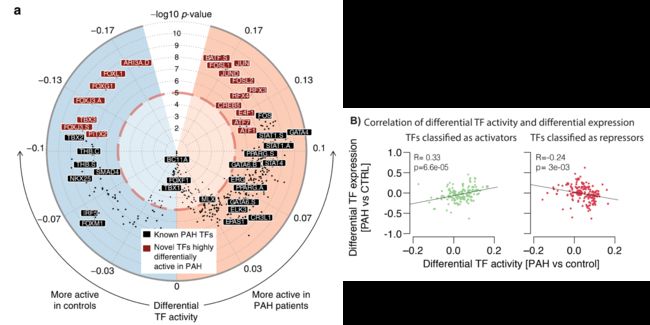
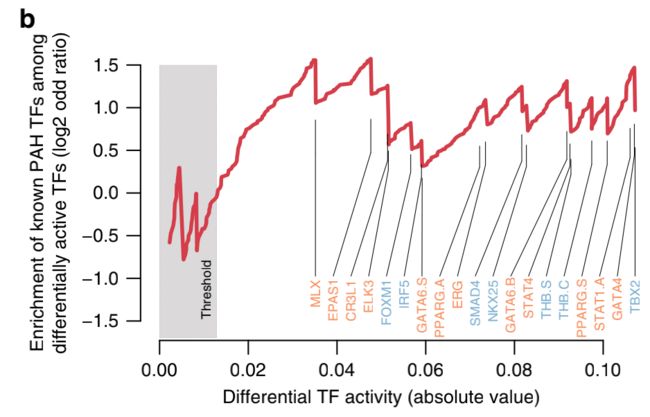
总而言之,在之前与PAH相关的20个TF中(编译自https://monarchinitiative.org, https:// www.opentargets.org/, and ref. 15见方法;补充数据4),16个被鉴定为差异活性TF,并且它们在差异活性TF中被强烈富集,而不管用于定义差异活性TF的阈值如何(图3b)。
这表明我们评估TF活性的方法对于识别潜在的重要疾病驱动因素TF是可靠且灵敏的。SOX17,最近才被确认为PAH相关TF[7,16]在我们的PAH患者中也有显著的缺失调节(补充数据4)。
一些TF(17个代表18个基序;图3a)显示出比已知的PAH-TF更强的PAH和对照之间的差异活性,这表明它们可能是导致该疾病的新候选调控因子。这些新发现的TF包括AP1复合体(BATF、FOSL1、JUN、JUND、FOSL2以及ATF1和ATF7,它们都有相似的结合位点,因此应该被认为是无法区分的),RFX家族的成员(RFX3、RFX4,又是相似的基序),CREB5和E4F1在患者中活性更高,以及ARID3A,FOX家族的成员(FOXL1,FOXG1,FOXJ3,也是类似的。
AP1复合体是一种已知的促炎因子[17],鉴于作者以往的发现,炎症在PAH[18,19]中扮演着重要的角色,因此特别令人感兴趣。类似地,E4F1和ARID3A已被证明与p53[20,21]的特定功能相互作用与激活,最近作者研究发现,在与炎症相关的过程中,当缺失BMPR2时,p53特异性上调。CREB5已被认为是肺组织中的一个缺氧反应基因[23],其活性增强可能是疾病marker。RFX家族的转铁蛋白长期以来被认为与胶原的抑制有关[24],而胶原又是一个已知的PAH相关基因(补充数据4)。最后,TBX4与TBX3具有高度相似的结合位点,其突变与儿童期发病的PAH有关[25]。这些例子表明,许多新预测的PAH-TF可能确实在发病机制中发挥了作用,并为进一步研究提供了重要的研究基础。
4.Gene regulatory network and PAH-specific TFs highlight PAH biology.
下一步作者想要研究在PAH中受不同活性TF调控的靶基因。
为此,作者试图建立一个PAH特异的基因调控网络,将TF与它们的靶基因联系起来。
前面,作者已经定义了基因和它们的增强子之间的联系(图2c),所以现在只需要把TF与它们调节的调控元件联系起来。
为此,作者设计了一种基于相关性的方法,如果(I)调控元件具有假定的TF结合位点,并且(II)与TF的表达水平显著相关(正或负,反映激活因子和抑制因子),则将TF与其靶调控元件联系起来(图4a)。
这假设TF表达的任何变化都应该导致它正在调控的增强子和启动子的活性的变化。
注意,该方法基于不同个体的TF表达和增强子信号的协变,并且与条件之间的差异表达/信号无关。
为了估计TF-peak的FDR,我们使用TF与不包含其motifs的峰的相关性作为背景来计算经验FDR。我们选择20%的FDR作为界限,因为这似乎可以最大化“一致”交互的比例,从而一致被定义为TF的活动和它的目标peak沿着相同的方向进行的交互(图4b)。值得注意的是,我们观察到<20%的增强子与邻近的基因相关(就到TSS的距离而言),这再次强调了细胞类型特异的基因-增强子互作的重要性(图4c)。
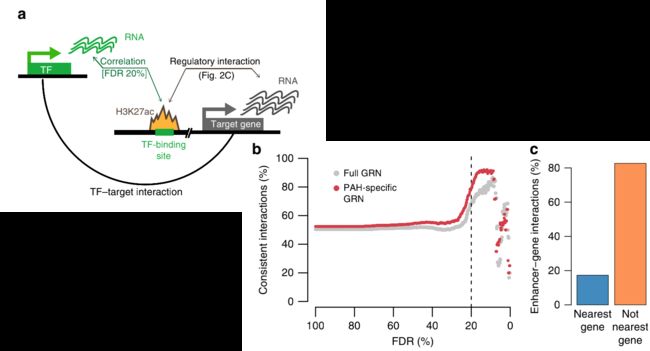
为了专注于解决疾病特异性的相互作用,作者选择了由上面获得的假定的PAH-TFs调控的子网络。一个PAH特异的基因调控网络,包括116个TFs,1880个目标基因和5342个相互作用网络(图4d-f,补充数据5)。为了给网络分配方向性,作者使用了基于Motif的diffTF计算的TF活性,还使用了CHIP-seq数据(参见方法)。TFs在基序和芯片序列数据之间的方向性不一致,但在至少一项分析中具有显著性,被标记为“混合证据”。然后,这个网络被用来查询特定基因集合之间的调控关系。
作为第一个例子,我们可视化验证了涉及先前已知的PAH相关TF的调控相互作用(图4g,补充数据4)。
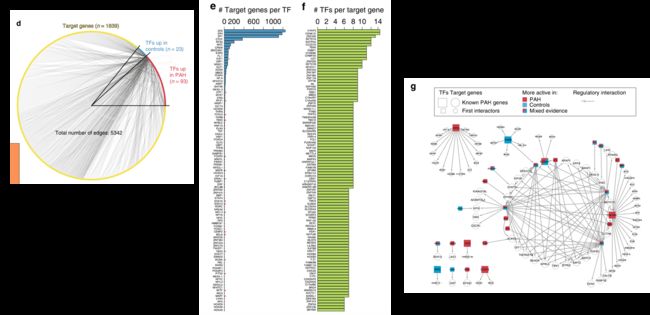
在306个已知的PAH相关基因中,有209个在我们的数据中表达,其中我们的GRN覆盖了9个TF和45个靶基因。
最后作者发现这些基因群分为一个由GATA6和SP1、SP2和SP4主导的主要调控簇,以及受STAT4、ERG和THRB等调控的几个较小的调控簇。
这表明,虽然有不同的调控途径受到影响,但许多已知的PAH基因参与了同一级联调控。
为了评估基因调控网络的生物学意义,作者对目的基因进行了GO富集分析,以所有表达的基因为背景,排除了TF。
我们发现超过250个GO terms在目标基因中显著富集(补充数据6;图5a中显示的子集)。
许多富集的生物学过程与内皮生物学有关,也与PAH有关。包括细胞迁移和管状形态发生(“regulation of cell migration” (OR = 1.4, adj. p-val = 4.1e-3) and “regulation of tube size” (OR = 2.5, adj. p-val = 0.011;补充数据6)),它们在PAH来源的内皮细胞中是下调的。
其他通路,如“细胞对皮质类固醇刺激的反应”(OR = 3.2, adj. p-val = 0.02)和“血管收缩调节”(OR = 3.7, adj. p-val = 0.016)可能是疾病和患者在肺移植前接受的治疗的结果,因为所有患者都接受了某种血管扩张剂药物的治疗(图5a,补充数据1)。
随后作者确定的最具体的GO是“SMAD蛋白复合物assembly”,这一过程受到导致HPAH的BMPR2突变的影响(图5B)。
总体而言,这些terms表明,我们的调控网络,由PAH患者不同活性的TF驱动,能够恢复预期的生物学过程,从而验证了该方法。
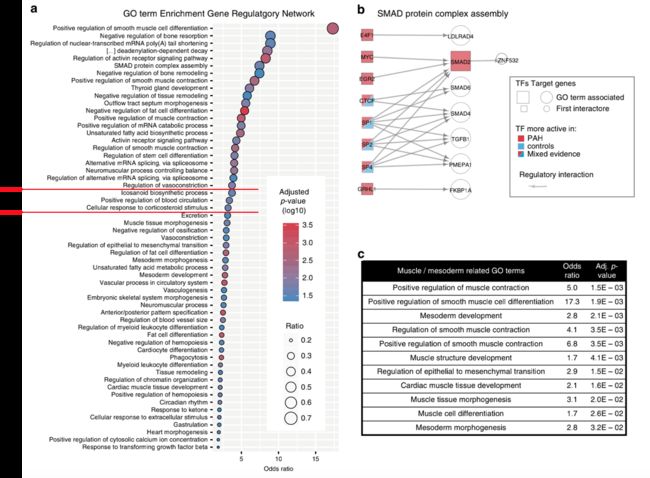
接下来,作者使用PAH调控网络来揭示疾病发生机制的过程。在PAH特异性调控网络中,最富集的GO term是“平滑肌细胞分化的正调控(positive regulation of smooth muscle cell differentiation)”,为此,作者确定了10个基因中的7个(OR = 17, adj. p-val = 1.9e-3;图5a,c)。
他们还发现了其他与肌肉生物学相关的生物过程,如“上皮向间充质转化”和“中胚层发育”,这些都是富集程度较高的通路(图5c)。向GRN查询这些GO中TF活性的方向性,发现大多数调节它们的TF在PAH中更活跃,从而表明这些通路在PAH中上调。随着“细胞增殖”的增加(OR = 1.4, adj. p-val = 4.9e-3)这可能提示增殖的内皮细胞开始分化为平滑肌样细胞的机制。这是如何发生的一个线索来自term“对转化生长因子β的反应”(OR = 1.8, adj. p-val = 0.045)。提示内皮细胞对生长因子刺激的异常反应可能导致细胞增殖和EndMT,并可能最终促进PAH中典型的平滑肌细胞过度增殖[26]。调控网络表明,转化生长因子家族TGFβ1,也包括在平滑肌分化GO term中,受SP家族(SP1, SP2, SP4)的TF控制,这些TF在肺动脉高压患者中具有不同的活性(图5d)。更重要的是,作者发现通过TGFβ激活的SMAD2 TF在肺动脉高压中具有很强的活性(图5e),因此提示肺动脉高压患者内皮细胞中TGFβ通路的调控缺失与疾病发生相关。它还提示与炎症(SP家族)、信号转导(SMADS)和EndMT(SNAI1)相关的TF之间存在以前未知的联系。
总之,我们的数据表明,肺动脉高压患者活跃的染色质景观在调节平滑肌细胞分化相关基因的增强子上发生了特异性的改变,并可能对TGFβ信号做出异常反应。这也解释了RNA和H3K27ac标记之间缺乏相关性的原因。因此,我们预测,在内皮生长或信号因子的刺激下,这些靶基因中的一些在PAH患者和对照组中的表达反应将显著不同。

5.RNA response to growth factor reflects steady state H3K27ac.
为了验证PAH患者PAECs中的增强子在启动特定基因以对生长因子刺激做出异常反应的假设,作者在一组预测在PAH患者中被不同启动的基因中,测量了PAECs中相关的三个刺激的反应。这些基因被选择来代表发现富集在PAH GRN中的不同过程,并且需要(在作者的GRN中)连接到一个不同的H3K27-acetylated peak。
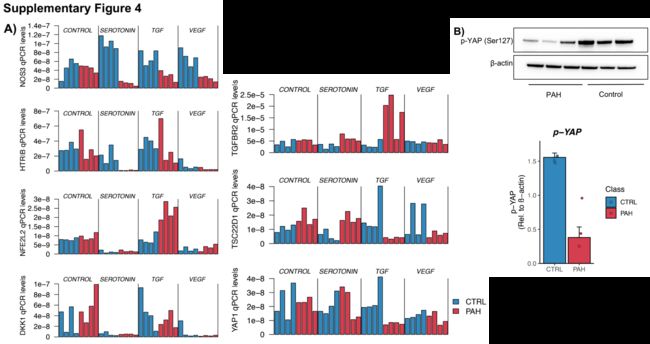
具体地说,我们选择了NOS3和HTR1B(血管系统),TSC22D1(基本转录过程),YAP1和DKK1(中胚层发育),NFE2L2(刺激反应)和TGFβ受体2(TGFβ的直接靶标)。
我们使用了8个肺动脉内皮细胞株-4个来自患者(2个IPAH和2个HPAH)和4个来自对照组-并使它们受到三种与内皮细胞相关的不同生长因子/信号分子的刺激:血管内皮生长因子(VEGF10 ng/ml)、转化生长因子β(TGFβ;10 ng/ml)和5-羟色胺(serotonin,10uM),每种刺激24小时,然后我们对选定的基因进行qPCR,比较PAH和对照组的表达。
正如从全局RNA-Seq数据中结果的那样,在未刺激的肺动脉内皮细胞中,患者和对照组之间没有检测到显着变化(图6a)。
然而,我们发现大多数基因在至少一种刺激下在患者和对照组之间表现出显著的差异(补充图4A)。
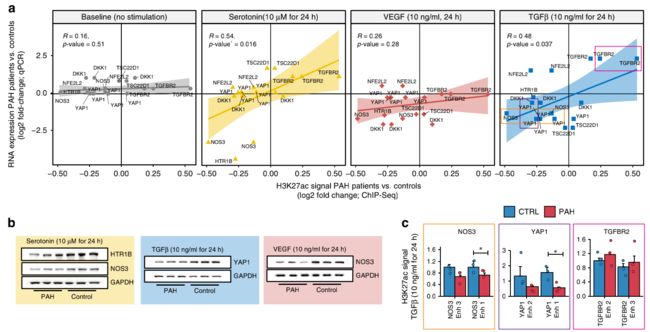
重要的是,当比较PAH和对照组时,我们发现基线时增强子活性(H3K27ac)的变化与内皮信号下其靶基因表达的变化之间存在很强的相关性(图6a)。
这证实了这样的观点,即这些基因正被增强子激活,在PAH患者中以异常的方式做出反应,从而可能导致异常生长或分化。对于TGF-β刺激,我们发现差异调控基因的增强子在刺激时变得异常活跃(图6b)。
为了扩大这些观察到的功能重要性,作者的结果还表明,在每种刺激中,一些基因的蛋白质水平(用WB测量)与mRNA水平一致(图6c)。对于受磷酸化高度调控的YAP1,作者还通过实验验证了磷酸化形式根据其RNA水平的变化(补充图4B)。
下面的例子可以作为在PAH疾病机制背景下解释数据的概念的证明:
首先,VEGF通常诱导NOS3[27]。在PAH-PAECs中,其增强子与对照-PAECs相比H3K27ac信号减少了。
同时NOS3 mRNA和蛋白对VEGF的反应仅在对照组肺动脉内皮细胞中增强。
其次,5-羟色胺通常通过HTR1B受体C(HTR1BRC)刺激血管生成。
然而,在PAH-PAEC中,HTR1BRC的增强子显示H3K27ac信号减弱,似乎可以使细胞对抗5-羟色胺刺激后HTR1BRC mRNA和蛋白的诱导,从而提供了另一种破坏PAH血管生成的方式,这与5-羟色胺反应中NOS3的减少是一致的。或者,NOS3增强剂子H3K27ac标记的减少可能会改变其在PAH中的反应。
第三,YAP1促进内皮细胞的血管生成[28],但是YAP1在内皮细胞中不受TGFβ的调节[29]。在此,TGFβ对肺动脉高压肺动脉内皮细胞YAP1和NOS3表达的上调也可能是这些细胞血管生成受损的原因。
最后,新的TF,如NFE2L2和TSC22D1,似乎被TGFβ异常上调,可能与干扰这一途径的新机制有关。因此,H3K27ac标记帮助我们发现在内皮再生中重要的新通路和效应器,以响应在疾病(如肺动脉高压)中受损的刺激。
总而言之,这些实验验证了作者的预测,即PAH患者的内皮细胞对生长因子刺激的反应方式与健康对照组明显不同,这一机制可能涉及增强子启动。
综上所述,本研究结果为肺动脉高压的表观遗传学基础提供了AP1、TGFβ、5-羟色胺和VEGF信号的异常,这些信号共同导致炎症、异常的血管生成和EndMT,以及平滑肌细胞增殖的倾向,这些都是肺动脉高压的特征。此外,结果表明,潜在的治疗方法不仅应该考虑它们对细胞表面受体或信号通路的影响,而且应该考虑它们是否逆转了异常的表观遗传的影响。
小结
环境和表观遗传因素往往在多基因疾病中起重要作用。
作者从患者和对照组(n=19)的肺组织中获得肺动脉内皮细胞(PAECs),并进行染色质、转录和相互作用分析。
作者观察到PAH-PAEC中活性增强子的广泛重构,并鉴定了数百个不同活性的TF,但在稳定状态下发现的转录变化很小。
设计了一个疾病特异性的增强子-基因调控网络,并预测PAH患者PAECs中的启动增强子被不同活性的TF激活,导致其对内皮信号的异常反应,这可能导致血管生成障碍和内皮到间质的转变。
在体外实验验证了在转化生长因子-β、血管内皮生长因子或5-羟色胺刺激的肺动脉内皮细胞中选择靶基因的预测。
本研究强调了染色质状态和活性增强子在疾病相关的PAH细胞类型中的作用。
讨论
本研究表明,PAH患者的小肺动脉内皮细胞(PAECs)在稳态时表现出增强子的重塑,但没有显示任何显著差异表达的基因。这一明显的矛盾使作者提出科学假设:增强子可能启动PAH中的PAECs,使其对内皮信号做出异常反应,作者在实验上通过一些基因证实了这一点。
使用染色质构象和基于相关性的方法将基因与其近端和远端的调控元件联系起来,作者发现了特异的内皮和PAH相关过程,表明PAH中的PAEC在正常的内皮信号作用下向内皮到间质的转变启动。这突出了将基因与其远端调控元件联系起来以揭示生物功能的重要性。
通过使用diffTF[8]将H3K27ac信号转换为活性TF分布,我们在患者和对照组之间识别出了大量不同活性的TF,包括几乎所有先前已知的PAH相关TF。
结合RNA, ChIA-PET和H3K27ac数据,作者设计了一种方法来生成基因调控网络,该网络能够捕获与疾病相关的生物学过程。除了生物学上的关联,这也表明(a)从供体肺中获取并培养了几代的细胞保持其调控状态,潜在地编码为表观遗传记忆,以及(b)本研究中的调控网络方法能够捕捉到重要的生物学功能。
作者注意到,其构建的基因调控网络是基于TF和调控元件之间的相互作用,这些元件在不同的个体之间是不同的。
因此,我们不太可能找回高度保守和严格调控的过程,而是那些可能因个体而异的过程,因此可能在聚集在导致疾病的分子途径上发挥作用。同时还注意到,使用CTCF-CHIA-PET作为调用染色质调节域的基础的一个缺陷是,将对任何没有被CTCF划定的基因loop失去观察力。
通过使用疾病特异性转录因子探索这个调控网络,作者在top GO term中确定了与平滑肌细胞分化相关的过程。这一点特别有趣,因为作者之前的发现表明,PAEC中BMPR2活性的降低促进了EndMT[26]。然而,这一先前的发现是对BMPR2下调的特异性反应,并由HMGA1和Slug介导。
在本次研究的网络分析中,作者发现,overall regulatory state可能会推动细胞走向EndMT。这一点特别有趣,因为这次并没有发现BMPR2的显著下调。Slug和HMGA1是我们网络中的靶基因,可能有助于EndMT作为PAH过程的富集。
最后,尽管没有发现任何差异表达的基因,PAECs中的H3K27ac水平对选择的一组基因的内皮信号因子的表达变化具有很高的预测性。
这表明,虽然这些从肺动脉最内皮层分离的细胞仍然具有内皮命运,但它们确实显示出向间充质转化为平滑肌细胞的启动。
这种转变可能是由炎症以及5-羟色胺和TGFβ等信号分子触发的。EndMT导致抑制平滑肌细胞增殖的内皮因子缺失,从而导致PAH患者增生的平滑肌细胞扩张的新生内膜堵塞血管腔。
总而言之,本研究提出了一个大的框架,用于在增强子介导的基因网络中集成多个组学数据,允许识别驱动疾病特定途径的基因,并对疾病机制提供见解。
它表明,稳态表达分析仅限于机制涉及对刺激的异常反应的系统。考虑到增强子,特别是活性标记H3K27ac的增强子,可能会导致仅靠基因表达或启动子分析无法获得的见解。看看H3K27ac标记的增强子是否可以在其他系统中充当启动因子,从而在某种程度上为稳态研究增加了一个更动态的视角,这将是一个有趣的事情。
Data availability
The raw sequencing data are available at the Gene Expression Omnibus (GEO) data repository under the accession number GSE126325. Chromatin modififications profifiling data have been deposited under the accession number GSE126322. RNA-Seq data have been deposited under the accession number GSE126262. Chromatin interaction profifiling (ChIA-PET) data mediated by CTCF have been deposited under the accession number GSE139234. The source data underlying Figs. 1f, 2a, b, d, e, h, 6c and Supplementary Figs. 2a, 4a, b are provided as a Source Data fifile.
Code availability
Code is available upon request from the corresponding author [email protected].
Received: 4 March 2019; Accepted: 13 March 2020;
References
\1. Morrell, N. W. et al. Genetics and genomics of pulmonary arterial
hypertension. Eur. Respir. J. 53, 1801899 (2019).
\2. Lau, E. M. T., Giannoulatou, E., Celermajer, D. S. & Humbert, M.
Epidemiology and treatment of pulmonary arterial hypertension. Nat. Rev.
Cardiol. 14, 603–614 (2017).
\3. Benza, R. L. et al. An evaluation of long-term survival from time of diagnosis
in pulmonary arterial hypertension from the REVEAL Registry. Chest 142,
448–456 (2012).
\4. Rabinovitch, M. Molecular pathogenesis of pulmonary arterial hypertension. J.
Clin. Invest. 122, 4306–4313 (2012).
\5. Simonneau, G. et al. Updated clinical classifification of pulmonary
hypertension. J. Am. Coll. Cardiol. 62, D34–D41 (2013).
\6. Machado, R. D. et al. Pulmonary arterial hypertension: a current perspective
on established and emerging molecular genetic defects. Hum. Mutat. 36,
1113–1127 (2015).
\7. Gräf, S. et al. Identifification of rare sequence variation underlying heritable
pulmonary arterial hypertension. Nat. Commun. 9, 1416 (2018).
\8. Berest, I. et al. Quantifification of differential transcription factor activity and
multiomics-based classifification into activators and repressors: diffTF. Cell Rep.
29, 3147–3159 (2019). e12.
\9. Rhodes, C. J. et al. RNA sequencing analysis detection of a novel pathway of
endothelial dysfunction in pulmonary arterial hypertension. Am. J. Respir.
Crit. Care Med. 192, 356–366 (2015).
\10. Sa, S. et al. Induced pluripotent stem cell model of pulmonary arterial
hypertension reveals novel gene expression and patient specifificity. Am. J.
Respir. Crit. Care Med. 195, 930–941 (2017).
\11. Gu, M. et al. Patient-specifific iPSC-derived endothelial cells uncover pathways
that protect against pulmonary hypertension in BMPR2 mutation carriers.
Cell Stem Cell 20, 490–504 (2017).
\12. Roadmap Epigenomics, Consortium et al. Integrative analysis of 111 reference
human epigenomes. Nature 518, 317–330 (2015).
\13. Delaneau, O. et al. Chromatin three-dimensional interactions mediate genetic
effects on gene expression. Science 364, eaat8266 (2019).
\14. Grubert, F. et al. Genetic control of chromatin states in humans involves local
and distal chromosomal interactions. Cell 162, 1051–1065 (2015).
\15. Garcia-Rivas, G., Jerjes-Sánchez, C., Rodriguez, D., Garcia-Pelaez, J. &
Trevino, V. A systematic review of genetic mutations in pulmonary arterial
hypertension. BMC Med. Genet. 18, 82 (2017).
\16. Rhodes, C. J. et al. Genetic determinants of risk in pulmonary arterial
hypertension: international genome-wide association studies and meta�
analysis. Lancet Respir. Med. 7, 227–238 (2019).
\17. Angel, P. & Karin, M. The role of Jun, Fos and the AP-1 complex in cell�
proliferation and transformation. Biochim. Biophys. Acta 1072, 129–157
(1991).
\18. Rabinovitch, M., Guignabert, C., Humbert, M. & Nicolls, M. R. Inflflammation
and immunity in the pathogenesis of pulmonary arterial hypertension. Circ.
Res. 115, 165–175 (2014).
\19. Boucherat, O. et al. The cancer theory of pulmonary arterial hypertension.
Pulm. Circ. 7, 285–299 (2017).
\20. Le Cam, L. et al. E4F1 is an atypical ubiquitin ligase that modulates p53
effector functions independently of degradation. Cell 127, 775–788 (2006).
\21. Lestari, W. et al. Cooperation between ARID3A and p53 in the transcriptional
activation of p21WAF1 in response to DNA damage. Biochem. Biophys. Res.
Commun. 417, 710–716 (2012).
\22. Diebold, I. et al. BMPR2 preserves mitochondrial function and DNA during
reoxygenation to promote endothelial cell survival and reverse pulmonary
hypertension. Cell Metab. 21, 596–608 (2015).
\23. Leonard, M. O. et al. Hypoxia selectively activates the CREB family of
transcription factors in the in vivo lung. Am. J. Respir. Crit. Care Med. 178,
977–983 (2008).
\24. Ponticos, M. & Smith, B. D. Extracellular matrix synthesis in vascular disease:
hypertension, and atherosclerosis. J. Biomed. Res. 28, 25–39 (2014).
\25. Kerstjens-Frederikse, W. S. et al. TBX4 mutations (small patella syndrome) are
associated with childhood-onset pulmonary arterial hypertension. J. Med.
Genet. 50, 500–506 (2013).
\26. Hopper, R. K. et al. In pulmonary arterial hypertension, reduced BMPR2
promotes endothelial-to-mesenchymal transition via HMGA1 and its target
slug. Circulation 133, 1783–1794 (2016).
\27. Chen, Y., Medhora, M., Falck, J. R., Pritchard, K. A. & Jacobs, E. R.
Mechanisms of activation of eNOS by 20-HETE and VEGF in bovine
pulmonary artery endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 291,
L378–L385 (2006).
\28. He, J. et al. Yes-associated protein promotes angiogenesis via signal transducer
and activator of transcription 3 in endothelial cells. Circ. Res. 122, 591–605
(2018).
\29. Shoeibi, S., Mozdziak, P. & Mohammadi, S. Important signals regulating
coronary artery angiogenesis. Microvasc. Res. 117, 1–9 (2018).
\30. Mahler, G. J., Farrar, E. J. & Butcher, J. T. Inflflammatory cytokines promote
mesenchymal transformation in embryonic and adult valve endothelial cells.
Arterioscler. Thromb. Vasc. Biol. 33, 121–130 (2013).
\31. Kasowski, M. et al. Extensive variation in chromatin states across humans.
Science 342, 750–752 (2013).
\32. Landt, S. G. et al. ChIP-seq guidelines and practices of the ENCODE and
modENCODE consortia. Genome Res. 22, 1813–1831 (2012).
\33. Fullwood, M. J., Han, Y., Wei, C.-L., Ruan, X. & Ruan, Y. Chromatin
interaction analysis using paired-end tag sequencing. Curr. Protoc. Mol. Biol.
Chapter 21, 1–25 (2010).
\34. Heidari, N. et al. Genome-wide map of regulatory interactions in the human
genome. Genome Res. 24, 1905–1917 (2014).
\35. Phanstiel, D. H., Boyle, A. P., Heidari, N. & Snyder, M. P. Mango: a bias�
correcting ChIA-PET analysis pipeline. Bioinformatics 31, 3092–3098 (2015).
\36. Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and
dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
\37. Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flflexible trimmer for
Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
\38. Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows�
Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
\39. Carroll, T. S., Liang, Z., Salama, R., Stark, R. & de Santiago, I. Impact of
artifact removal on ChIP quality metrics in ChIP-seq and ChIP-exo data.
Front. Genet. 5, 75 (2014).
\40. Lun, A. T. L. & Smyth, G. K. csaw: a Bioconductor package for differential
binding analysis of ChIP-seq data using sliding windows. Nucleic Acids Res.
44, e45 (2016).
\41. Kulakovskiy, I. V. et al. HOCOMOCO: towards a complete collection of
transcription factor binding models for human and mouse via large-scale
ChIP-Seq analysis. Nucleic Acids Res. 46, D252–D259 (2018).
\42. Gheorghe, M. et al. A map of direct TF-DNA interactions in the human
genome. Nucleic Acids Res. 47, e21 (2019).