- 【DeepSeek】复现DeepSeek R1?快来看这个Open R1项目实践指南~
FF-Studio
DeepSeekR1语言模型自然语言处理深度学习人工智能
OpenR1项目基于DeepSeek-R1的技术报告和方法论,公开并复现R1的训练管线,并且希望所有开发者都能在这个基础上搭建自己的研究或应用。笔者研读了大量资料,对OpenR1的愿景、原理及在实践层面的具体操作,产生了许多想法。因此,这篇博客会从最初的概念入手,带领大家了解OpenR1的原理与技术细节,并侧重讲解其中最为关键的强化学习训练方法之一——GRPO(群组相对策略优化,GroupRela
- AI编译器之——为什么大模型需要Relax?
FF-Studio
人工智能深度学习自然语言处理机器学习语言模型
放在最前:Relax的关键创新深度学习模型(比如ChatGPT这种大模型)在运行时经常遇到“输入尺寸不固定”的情况。比如你问它一个问题,这次输入是10个字,下次可能是100个字。传统编译器处理这种“变来变去”的尺寸很笨——要么只能按固定尺寸优化(导致变尺寸时性能暴跌),要么每次都要重新编译(慢到没法用)。Relax的创新:符号形状:让编译器学会“代数”Relax允许编译器用“符号变量”(比如n)表
- 实战LLM强化学习——使用GRPO(DeepSeek R1出圈算法)
FF-Studio
DeepSeekR1算法语言模型人工智能自然语言处理机器学习
——关于使用Unsloth库、LoRa微调及GRPOTrainer自定义奖励函数实现“只输出10个英语单词”的探索为什么要进行“只输出10个英文单词”的极端尝试?在大模型的训练或微调当中,大多数场景我们都希望它能“自由发挥”,给出越丰富越好的答案。但,为了更好的理解强化学习在LLM训练过程中发挥的意义,也为了学习GPRO这个强化学习算法,笔者出此题目,方便大家学习理解。GRPO(GroupRela
- Ubuntu交叉编译 arm板子上的TVM
陈有爱
TVMubuntu人工智能
目录X86Ubuntu的TVM安装LLVM下载tvm配置config.cmake编译源码python安装测试是否安装成功可以在安装一些库,用于RPCTracker和auto-tuning交叉编译801arm的TVM交叉编译链下载配置config.cmake编译源码编译的时候可能会遇到错误ONNX模型转换为TVM模型创建pre.py,将onnx模型编译成tvm.so文件测试TVM模型修改demo程序
- Android Room 使用
francisHuang
android学习androidRoom数据库
官网介绍:https://developer.android.google.cn/training/data-storage/roomRoom是在SQLite上提供了一个抽象层,以便在充分利用SQLite的强大功能的同时,能够流畅地访问数据库。Room包含3个重要部分:数据库:包含数据库持有者,并作为应用已保留的持久关系型数据的底层连接的主要接入点。Entity:表示数据库中的表。DAO:包含用于
- 【Python入门基础】——第1篇:从入门到精通:Python简介与环境搭建详解
猿享天开
python从入门到精通python开发语言
第1篇:Python简介与环境搭建目录什么是Python?Python的历史与特点安装Python解释器配置开发环境选择合适的集成开发环境(IDE)使用文本编辑器运行第一个Python程序常见问题及解决方法总结什么是Python?Python是一种高级、通用、解释型的编程语言,由GuidovanRossum于1991年首次发布。Python以其简洁易读的语法、广泛的应用领域和强大的社区支持,成为全
- 【2024年-12月-续篇-开源社区openEuler实践记录】go-from-mod
我明天再来学Web渗透
开源社区OpenEuler开源golang开发语言copilot架构开源软件后端
前言初学Go语言,下面仅为个人所学以及小结,若有错误之处,还请指教。Go语言基础入门篇的一二节课,其中我对每个讲到的语法都重写或者本地测试运行过,其中第二节课的第三个小项目尚未实现(本人对网络连接那块的脚本尚不熟悉,)部分代码不能太多,所以贴图了有些。Go基础语法目前学过的Go语法只有课程内的,下面是一些小小的笔记。导包和输出packagemainimport"fmt"funcmain(){fmt
- deepseek v1手机端部署
哎呀——哪是啥
智能手机
在iPhone上部署DeepSeekR11.安装快捷指令:打开iPhone上的Safari浏览器,访问[这个链接](https://www.icloud.com/shortcuts/e0bc5445c39d45a78b90e1dc896cd010)下载快捷指令。下载后,按照提示完成安装。2.获取并配置APIKey:访问[这个链接](https://dev.hkgpt.top/shop/46)获取你
- YOLOv11-ultralytics-8.3.67部分代码阅读笔记-head.py
红色的山茶花
YOLO笔记深度学习
head.pyultralytics\nn\modules\head.py目录head.py1.所需的库和模块2.classDetect(nn.Module):3.classSegment(Detect):4.classOBB(Detect):5.classPose(Detect):6.classClassify(nn.Module):7.classWorldDetect(Detect):8.cl
- Vue2之 v-if VS v-show
问老大
前端javascript开发语言vue.js
Vue2中的v-if和v-show都是用来实现条件性渲染的指令,用于控制元素显示与隐藏的指令,但它们在实现机制和使用场景上有所不同:一、实现机制:1.1、v-if当条件表达式为真时,Vue.js会根据条件动态地创建或销毁对应的DOM元素。当条件为假时,Vue.js会销毁对应的DOM元素,并且从DOM中移除。每次条件改变时,Vue.js都会重新进行DOM的创建或销毁,这可能会导致性能开销较高,尤其是
- python与excel整合全教程
刘同学Python学习日记
pythonexcel开发语言
Python与Excel的整合非常强大,尤其适合处理大数据、自动化表格操作以及进行高级数据分析。以下是一个全教程,涵盖常用的Python库及其应用:1.准备工作安装必要的库:使用以下命令安装常用库:pipinstallopenpyxlpandasxlrdxlsxwriterpywin32openpyxl:用于操作Excel的.xlsx文件(推荐)。pandas:强大的数据分析工具,支持读取和写入E
- 高效目录操作:如何使用 os.listdir 函数列出文件和文件夹
刘同学Python学习日记
学习记录os库python学习
在Python中,os.listdir()是一个用于列出指定目录下所有文件和子目录名称的函数。它来自于os模块,该模块提供了与操作系统进行交互的多种功能。importos#列出当前目录下的所有文件和子目录entries=os.listdir('.')print(entries)在这个示例中:os.listdir('.')将返回当前工作目录(用.表示)的所有文件和目录的名称列表。entries变量将
- 【AI中数学-数理统计-综合实例-包括python实现】 揭开数据的面纱:真实样本数据的探索与可视化
云博士的AI课堂
AI中的数学人工智能python数理统计数据预处理数据探索数据可视化机器学习
第五章:数理统计-综合实例1.揭开数据的面纱:真实样本数据的探索与可视化在人工智能(AI)应用中,数据是构建算法和模型的基石,而数理统计则为我们提供了理解和处理这些数据的工具。数据探索和可视化是数理统计中至关重要的步骤,它们不仅能帮助我们理解数据的分布、关系和趋势,还能够为后续的建模工作提供依据。本节将通过五个实际案例,展示如何使用数理统计和可视化技术对真实样本数据进行探索。每个案例都包括具体的描
- javascript使用字符串作为参数报错的问题
jumptigerfu
前端问题&解决方案elementui前端flaskjavascriptpython
原因:因为直接字符串作为参数需要加单引号,不然就判定为为定义的参数functionoption(value,row,index){varhtm="";htm+='导入'returnhtm;}解决方案:onclick里面加入转义字符\'functionoption(value,row,index){varhtm="";htm+='导入'returnhtm;}完美解决
- Vue 响应式渲染 - 模板语法
JSON_L
前端#Vuevue.js前端javascript
Vue渐进式JavaScript框架基于Vue2的学习笔记-Vue响应式渲染-模板语法目录模板语法渲染变量(状态)绑定事件简写事件修改属性样式修改绑定图片路径动态显示和隐藏总结模板语法渲染变量(状态)在页面中直接渲染变量。示例如下:Title{{myname}}newVue({el:"#box",//elementdata:{myname:'我的名字是张三'}})绑定事件增加按钮,并对按钮绑定点击
- 如何用主域名的子目录建立typecho博客
iamyzs
java阿里云
由于我已经有了一个主域名网站https://24365.online,打算在此主域名下建一个子目录作为博客网站站点。有人问了,为什么是在主域名下的子目录建博客,而不是采用一个单独的二级域名建博客呢?这是因为二级域名和主域名是2个网站,他们的seo自然也是分开的。而子目录是属于主域名网站的,因此是一个网站,seo只做一个网站的就好。这样两个网站劲往一起使,会相辅相成。1、首先要购买服务器。我打算用t
- 全国计算机一、二、三、四级考试备考资料
iamyzs
java
我整理了一些计算机等级考试的资料,大家有需要的拿去点击链接即可保存。参考链接:全国计算机一、二、三、四级考试备考资料-豌豆火博客01、全国计算机等级考试一二三四级笔试官方样卷02、计算机一级考试资料汇总(含17套真题+1000套选择题)03、计算机三级备考资料汇总(含数据库、网络、信息安全、嵌入式系统开发、Linux应用技术)04、计算机四级考试资料汇总((含数据库+网络+信息安全+嵌入式系统开发
- 全面解析:HTML页面的加载全过程(六)--浏览器渲染之分层 - Layer
huazi99于老师
html前端
分层原因在生成布局树之后,渲染进程会将一些复杂的3D动画、滚动条、高z-index的元素生成图层,并生成图层树交给GPU加速渲染。页面设计复杂,并且交互效果多。如不分层,用户的一个简单交互将导致整个页面的重新渲染,效率低下。分层好处通过分层,浏览器可以将复杂的页面元素分离成不同的图层,每个图层可以独立地进行渲染和更新,从而减少重排和重绘的次数。例如,当用户滚动页面时,只有可视区域的内容会被重新绘
- Python.NET 安装与使用教程
卫伊祺Ralph
Python.NET安装与使用教程项目地址:https://gitcode.com/gh_mirrors/py/pythonnet本教程将指导你了解并安装Python.NET——这是一个让Python程序员能够无缝集成.NET框架的开源库。1.项目目录结构及介绍在克隆或下载pythonnet的源代码仓库后,你会看到以下基本目录结构:pythonnet/├──LICENSE#许可文件├──MANIF
- Apache TVM:开源深度学习编译器栈的领跑者
计攀建Eliza
ApacheTVM:开源深度学习编译器栈的领跑者tvmOpendeeplearningcompilerstackforcpu,gpuandspecializedaccelerators项目地址:https://gitcode.com/gh_mirrors/tv/tvm项目介绍ApacheTVM是一个专为深度学习系统设计的编译器栈。它旨在弥合生产力导向的深度学习框架与性能和效率导向的硬件后端之间的差
- 可扩展性设计架构模式——开闭原则
goTsHgo
Java开闭原则java
1.概述在架构设计中,遵循开闭原则(Open/ClosedPrinciple,OCP),代码应该“对扩展开放,对修改关闭”是实现可扩展性的关键。这个原则指导我们设计系统时,应使其对新增功能开放,而对现有代码的修改封闭。这样,当系统需求变化或需要添加新功能时,我们可以通过添加新的代码模块而不是修改现有代码来实现,从而减少了对现有系统稳定性和已有功能的风险。底层原理解释开闭原则基于抽象构建架构。系统中
- Katana - 纯C语言实现的CSS解析器
蓬玮剑
Katana-纯C语言实现的CSS解析器katana-parserACSSparsinglibraryinpureC99项目地址:https://gitcode.com/gh_mirrors/ka/katana-parserKatana是一个纯C编写的库,用于解析CascadingStyleSheets(CSS)。它设计简洁,没有外部依赖,为其他工具和库(如linter、验证器、模板语言以及重构和
- 一个功能强大、操作易用的屏幕录制.Net开源工具 草稿箱
编程乐趣
c#.net开源
推荐一款免费开源的屏幕录制工具,凭借其强大的功能和用户友好的界面,受到非常多人喜欢!01项目简介该工具不仅支持全屏录制,还提供区域录制、游戏录制和摄像头录制等多种模式。不管是录制软件操作、游戏、直播、网络教学、课件制作还是在线视频,都可以满足你的需求。此外该工具还可以录制多种屏幕内容,如鼠标点击和键盘的输入等。02功能特色1、支持截屏功能;2、支持桌面、窗口、自定义区域录制;3、支持录制鼠标点击或
- 从0开始使用面对对象C语言搭建一个基于OLED的图形显示框架(绘图设备封装)
charlie114514191
OLED驱动开发记录单片机c语言学习嵌入式软件stm32OLED
目录图像层的底层抽象——绘图设备抽象如何抽象一个绘图设备?桥接绘图设备,特化为OLED设备题外话:设备的属性,与设计一个相似函数化简的通用办法使用函数指针来操作设备总结一下图像层的底层抽象——绘图设备抽象在上一篇博客中,我们完成了对设备层的抽象。现在,我们终于可以卖出雄心壮志的一步了!那就是尝试去完成一个最为基础的图形库。我们要做的,就是设计一个更加复杂的绘图设备。为什么是绘图设备呢?我们程序员都
- Apache Airflow 全面解析
由数入道
人工智能apacheAirflow
1.Airflow的定义与核心定位ApacheAirflow是一个开源的工作流自动化与调度平台,由Airbnb于2014年创建,2016年进入Apache孵化器,2019年成为顶级项目。其核心设计理念是“WorkflowsasCode”,通过编程方式定义、调度和监控复杂的数据流水线(Pipeline),适用于ETL、机器学习模型训练、数据湖管理、报表生成等场景。2.核心概念与架构解析2.1核心组件
- [Android]service命令的使用
aaajj
Androidandroid
在前面的讨论中,我们说到,如果在客户端懒得使用aidl文件生成的接口类进行binder,可以使用IBinder的transcat方法ParceldataParcel=Parcel.obtain();ParcelresultParcel=Parcel.obtain();dataParcel.writeInterfaceToken(DESCRIPTOR);//发起请求aProxyBinder.tran
- [Android]init中添加新的command
aaajj
Androidandroid
在Android的init进程中,command是用于定义启动时要执行的具体命令行指令的关键部分。init进程是Android系统启动的第一个进程,它负责初始化系统的各个组件,并启动必要的服务。command可以在init.rc文件及其包含的其他.rc文件中找到,通常作为on操作块或service定义的一部分。1.command的基本概念command是一条或多条具体的命令行指令,它们在特定条件下
- 数据结构与算法之排序: LeetCode 1356. 根据数字二进制下 1 的数目排序 (Ts版)
Wang's Blog
DataStructureandAlgorithms动态规划leetcode算法
根据数字二进制下1的数目排序https://leetcode.cn/problems/sort-integers-by-the-number-of-1-bits/description/描述给你一个整数数组arr。请你将数组中的元素按照其二进制表示中数字1的数目升序排序如果存在多个数字二进制中1的数目相同,则必须将它们按照数值大小升序排列请你返回排序后的数组示例1输入:arr=[0,1,2,3,4
- 使用STM32高级定时器通道和互补通道驱动有刷直流电机的单极性驱动程序
QoyOle
stm32单片机嵌入式硬件
在嵌入式系统中,有刷直流电机(BrushedDCMotor)是常见的驱动装置。为了有效地驱动有刷直流电机,我们可以利用STM32微控制器的高级定时器通道和互补通道。本文将详细介绍如何利用这些功能来实现有刷直流电机的单极性驱动,并提供相应的源代码。STM32微控制器提供了多个高级定时器,例如TIM1、TIM8等。这些高级定时器具有多个通道,每个通道可以用于产生PWM信号或输出高电平。在有刷直流电机的
- Oracle OCP证书,含金量到底有多高!
HCIE考证研究所
oracle开闭原则数据库网络工程师OCPOracle认证华为认证
很多小伙伴来了解OracleOCP证书,那本期就给大家详细讲解下,什么是Oracle?什么是OCP?01什么是Oracle认证?Oracle认证是由Oracle公司颁布并实施的一项权威认证,旨在满足对Oracle核心技术人才的需求。这项认证证明了个人在操作能力和广泛理论知识方面的专业水平。Oracle认证专为那些具备相关技能和知识的专业人士设计,以确保他们能够胜任使用、管理和支持Oracle产品的
- html
周华华
html
js
1,数组的排列
var arr=[1,4,234,43,52,];
for(var x=0;x<arr.length;x++){
for(var y=x-1;y<arr.length;y++){
if(arr[x]<arr[y]){
&
- 【Struts2 四】Struts2拦截器
bit1129
struts2拦截器
Struts2框架是基于拦截器实现的,可以对某个Action进行拦截,然后某些逻辑处理,拦截器相当于AOP里面的环绕通知,即在Action方法的执行之前和之后根据需要添加相应的逻辑。事实上,即使struts.xml没有任何关于拦截器的配置,Struts2也会为我们添加一组默认的拦截器,最常见的是,请求参数自动绑定到Action对应的字段上。
Struts2中自定义拦截器的步骤是:
- make:cc 命令未找到解决方法
daizj
linux命令未知make cc
安装rz sz程序时,报下面错误:
[root@slave2 src]# make posix
cc -O -DPOSIX -DMD=2 rz.c -o rz
make: cc:命令未找到
make: *** [posix] 错误 127
系统:centos 6.6
环境:虚拟机
错误原因:系统未安装gcc,这个是由于在安
- Oracle之Job应用
周凡杨
oracle job
最近写服务,服务上线后,需要写一个定时执行的SQL脚本,清理并更新数据库表里的数据,应用到了Oracle 的 Job的相关知识。在此总结一下。
一:查看相关job信息
1、相关视图
dba_jobs
all_jobs
user_jobs
dba_jobs_running 包含正在运行
- 多线程机制
朱辉辉33
多线程
转至http://blog.csdn.net/lj70024/archive/2010/04/06/5455790.aspx
程序、进程和线程:
程序是一段静态的代码,它是应用程序执行的蓝本。进程是程序的一次动态执行过程,它对应了从代码加载、执行至执行完毕的一个完整过程,这个过程也是进程本身从产生、发展至消亡的过程。线程是比进程更小的单位,一个进程执行过程中可以产生多个线程,每个线程有自身的
- web报表工具FineReport使用中遇到的常见报错及解决办法(一)
老A不折腾
web报表finereportjava报表报表工具
FineReport使用中遇到的常见报错及解决办法(一)
这里写点抛砖引玉,希望大家能把自己整理的问题及解决方法晾出来,Mark一下,利人利己。
出现问题先搜一下文档上有没有,再看看度娘有没有,再看看论坛有没有。有报错要看日志。下面简单罗列下常见的问题,大多文档上都有提到的。
1、address pool is full:
含义:地址池满,连接数超过并发数上
- mysql rpm安装后没有my.cnf
林鹤霄
没有my.cnf
Linux下用rpm包安装的MySQL是不会安装/etc/my.cnf文件的,
至于为什么没有这个文件而MySQL却也能正常启动和作用,在这儿有两个说法,
第一种说法,my.cnf只是MySQL启动时的一个参数文件,可以没有它,这时MySQL会用内置的默认参数启动,
第二种说法,MySQL在启动时自动使用/usr/share/mysql目录下的my-medium.cnf文件,这种说法仅限于r
- Kindle Fire HDX root并安装谷歌服务框架之后仍无法登陆谷歌账号的问题
aigo
root
原文:http://kindlefireforkid.com/how-to-setup-a-google-account-on-amazon-fire-tablet/
Step 4: Run ADB command from your PC
On the PC, you need install Amazon Fire ADB driver and instal
- javascript 中var提升的典型实例
alxw4616
JavaScript
// 刚刚在书上看到的一个小问题,很有意思.大家一起思考下吧
myname = 'global';
var fn = function () {
console.log(myname); // undefined
var myname = 'local';
console.log(myname); // local
};
fn()
// 上述代码实际上等同于以下代码
m
- 定时器和获取时间的使用
百合不是茶
时间的转换定时器
定时器:定时创建任务在游戏设计的时候用的比较多
Timer();定时器
TImerTask();Timer的子类 由 Timer 安排为一次执行或重复执行的任务。
定时器类Timer在java.util包中。使用时,先实例化,然后使用实例的schedule(TimerTask task, long delay)方法,设定
- JDK1.5 Queue
bijian1013
javathreadjava多线程Queue
JDK1.5 Queue
LinkedList:
LinkedList不是同步的。如果多个线程同时访问列表,而其中至少一个线程从结构上修改了该列表,则它必须 保持外部同步。(结构修改指添加或删除一个或多个元素的任何操作;仅设置元素的值不是结构修改。)这一般通过对自然封装该列表的对象进行同步操作来完成。如果不存在这样的对象,则应该使用 Collections.synchronizedList 方
- http认证原理和https
bijian1013
httphttps
一.基础介绍
在URL前加https://前缀表明是用SSL加密的。 你的电脑与服务器之间收发的信息传输将更加安全。
Web服务器启用SSL需要获得一个服务器证书并将该证书与要使用SSL的服务器绑定。
http和https使用的是完全不同的连接方式,用的端口也不一样,前者是80,后
- 【Java范型五】范型继承
bit1129
java
定义如下一个抽象的范型类,其中定义了两个范型参数,T1,T2
package com.tom.lang.generics;
public abstract class SuperGenerics<T1, T2> {
private T1 t1;
private T2 t2;
public abstract void doIt(T
- 【Nginx六】nginx.conf常用指令(Directive)
bit1129
Directive
1. worker_processes 8;
表示Nginx将启动8个工作者进程,通过ps -ef|grep nginx,会发现有8个Nginx Worker Process在运行
nobody 53879 118449 0 Apr22 ? 00:26:15 nginx: worker process
- lua 遍历Header头部
ronin47
lua header 遍历
local headers = ngx.req.get_headers()
ngx.say("headers begin", "<br/>")
ngx.say("Host : ", he
- java-32.通过交换a,b中的元素,使[序列a元素的和]与[序列b元素的和]之间的差最小(两数组的差最小)。
bylijinnan
java
import java.util.Arrays;
public class MinSumASumB {
/**
* Q32.有两个序列a,b,大小都为n,序列元素的值任意整数,无序.
*
* 要求:通过交换a,b中的元素,使[序列a元素的和]与[序列b元素的和]之间的差最小。
* 例如:
* int[] a = {100,99,98,1,2,3
- redis
开窍的石头
redis
在redis的redis.conf配置文件中找到# requirepass foobared
把它替换成requirepass 12356789 后边的12356789就是你的密码
打开redis客户端输入config get requirepass
返回
redis 127.0.0.1:6379> config get requirepass
1) "require
- [JAVA图像与图形]现有的GPU架构支持JAVA语言吗?
comsci
java语言
无论是opengl还是cuda,都是建立在C语言体系架构基础上的,在未来,图像图形处理业务快速发展,相关领域市场不断扩大的情况下,我们JAVA语言系统怎么从这么庞大,且还在不断扩大的市场上分到一块蛋糕,是值得每个JAVAER认真思考和行动的事情
- 安装ubuntu14.04登录后花屏了怎么办
cuiyadll
ubuntu
这个情况,一般属于显卡驱动问题。
可以先尝试安装显卡的官方闭源驱动。
按键盘三个键:CTRL + ALT + F1
进入终端,输入用户名和密码登录终端:
安装amd的显卡驱动
sudo
apt-get
install
fglrx
安装nvidia显卡驱动
sudo
ap
- SSL 与 数字证书 的基本概念和工作原理
darrenzhu
加密ssl证书密钥签名
SSL 与 数字证书 的基本概念和工作原理
http://www.linuxde.net/2012/03/8301.html
SSL握手协议的目的是或最终结果是让客户端和服务器拥有一个共同的密钥,握手协议本身是基于非对称加密机制的,之后就使用共同的密钥基于对称加密机制进行信息交换。
http://www.ibm.com/developerworks/cn/webspher
- Ubuntu设置ip的步骤
dcj3sjt126com
ubuntu
在单位的一台机器完全装了Ubuntu Server,但回家只能在XP上VM一个,装的时候网卡是DHCP的,用ifconfig查了一下ip是192.168.92.128,可以ping通。
转载不是错:
Ubuntu命令行修改网络配置方法
/etc/network/interfaces打开后里面可设置DHCP或手动设置静态ip。前面auto eth0,让网卡开机自动挂载.
1. 以D
- php包管理工具推荐
dcj3sjt126com
PHPComposer
http://www.phpcomposer.com/
Composer是 PHP 用来管理依赖(dependency)关系的工具。你可以在自己的项目中声明所依赖的外部工具库(libraries),Composer 会帮你安装这些依赖的库文件。
中文文档
入门指南
下载
安装包列表
Composer 中国镜像
- Gson使用四(TypeAdapter)
eksliang
jsongsonGson自定义转换器gsonTypeAdapter
转载请出自出处:http://eksliang.iteye.com/blog/2175595 一.概述
Gson的TypeAapter可以理解成自定义序列化和返序列化 二、应用场景举例
例如我们通常去注册时(那些外国网站),会让我们输入firstName,lastName,但是转到我们都
- JQM控件之Navbar和Tabs
gundumw100
htmlxmlcss
在JQM中使用导航栏Navbar是简单的。
只需要将data-role="navbar"赋给div即可:
<div data-role="navbar">
<ul>
<li><a href="#" class="ui-btn-active&qu
- 利用归并排序算法对大文件进行排序
iwindyforest
java归并排序大文件分治法Merge sort
归并排序算法介绍,请参照Wikipeida
zh.wikipedia.org/wiki/%E5%BD%92%E5%B9%B6%E6%8E%92%E5%BA%8F
基本思想:
大文件分割成行数相等的两个子文件,递归(归并排序)两个子文件,直到递归到分割成的子文件低于限制行数
低于限制行数的子文件直接排序
两个排序好的子文件归并到父文件
直到最后所有排序好的父文件归并到输入
- iOS UIWebView URL拦截
啸笑天
UIWebView
本文译者:candeladiao,原文:URL filtering for UIWebView on the iPhone说明:译者在做app开发时,因为页面的javascript文件比较大导致加载速度很慢,所以想把javascript文件打包在app里,当UIWebView需要加载该脚本时就从app本地读取,但UIWebView并不支持加载本地资源。最后从下文中找到了解决方法,第一次翻译,难免有
- 索引的碎片整理SQL语句
macroli
sql
SET NOCOUNT ON
DECLARE @tablename VARCHAR (128)
DECLARE @execstr VARCHAR (255)
DECLARE @objectid INT
DECLARE @indexid INT
DECLARE @frag DECIMAL
DECLARE @maxfrag DECIMAL
--设置最大允许的碎片数量,超过则对索引进行碎片
- Angularjs同步操作http请求with $promise
qiaolevip
每天进步一点点学习永无止境AngularJS纵观千象
// Define a factory
app.factory('profilePromise', ['$q', 'AccountService', function($q, AccountService) {
var deferred = $q.defer();
AccountService.getProfile().then(function(res) {
- hibernate联合查询问题
sxj19881213
sqlHibernateHQL联合查询
最近在用hibernate做项目,遇到了联合查询的问题,以及联合查询中的N+1问题。
针对无外键关联的联合查询,我做了HQL和SQL的实验,希望能帮助到大家。(我使用的版本是hibernate3.3.2)
1 几个常识:
(1)hql中的几种join查询,只有在外键关联、并且作了相应配置时才能使用。
(2)hql的默认查询策略,在进行联合查询时,会产
- struts2.xml
wuai
struts
<?xml version="1.0" encoding="UTF-8" ?>
<!DOCTYPE struts PUBLIC
"-//Apache Software Foundation//DTD Struts Configuration 2.3//EN"
"http://struts.apache
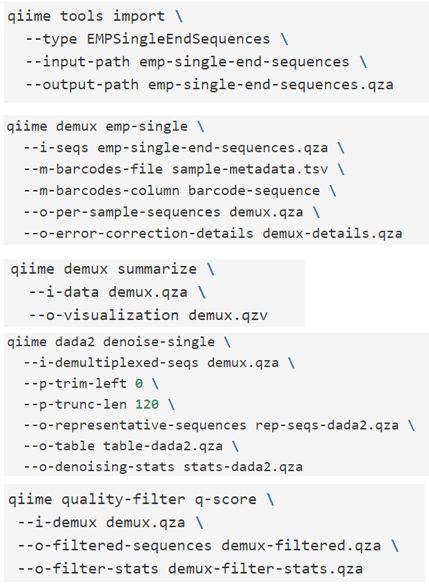
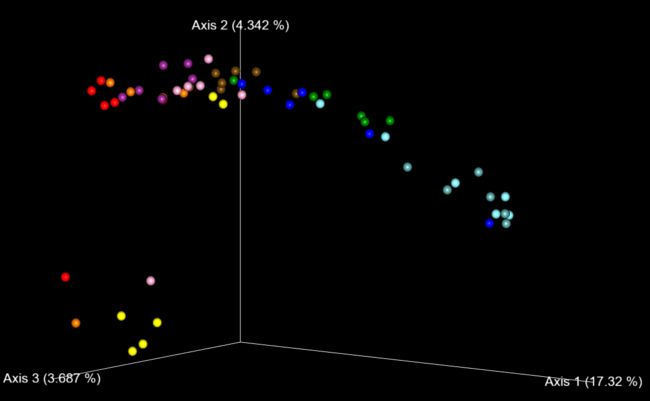


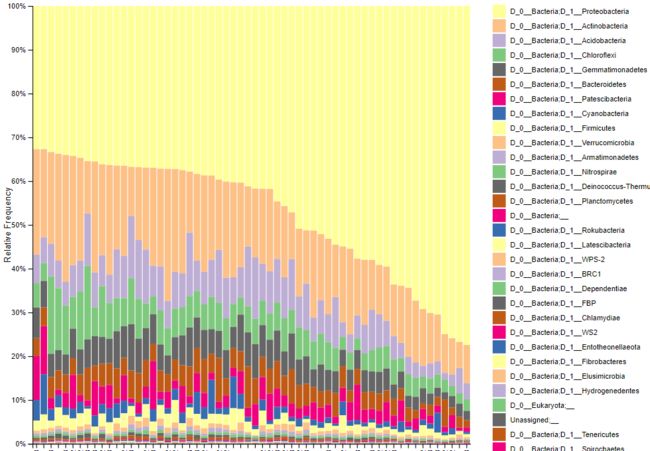
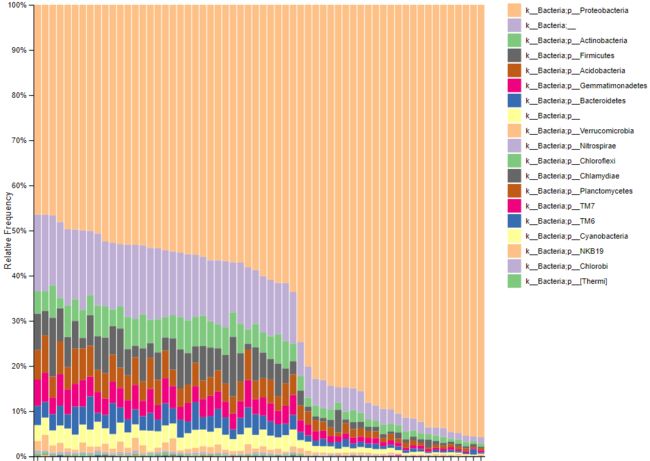
 image.png
image.png metadata
metadata