刘小泽写于2020.7.21
为何取名叫“交响乐”?因为单细胞分析就像一个大乐团,需要各个流程的协同配合
单细胞交响乐1-常用的数据结构SingleCellExperiment
单细胞交响乐2-scRNAseq从实验到下游简介
单细胞交响乐3-细胞质控
单细胞交响乐4-归一化
单细胞交响乐5-挑选高变化基因
单细胞交响乐6-降维
单细胞交响乐7-聚类分群
单细胞交响乐8-marker基因检测
单细胞交响乐9-细胞类型注释
单细胞交响乐9-细胞类型注释
单细胞交响乐10-数据集整合后的批次矫正
单细胞交响乐11-多样本间差异分析
单细胞交响乐12-检测Doublet
单细胞交响乐13-细胞周期推断
单细胞交响乐14-细胞轨迹推断
单细胞交响乐15-scRNA与蛋白丰度信息结合
单细胞交响乐16-处理大型数据
单细胞交响乐17-不同单细胞R包的数据格式相互转换
单细胞交响乐18-实战一 Smart-seq2
单细胞交响乐19-实战二 STRT-Seq
单细胞交响乐20-实战三 10X 未过滤的PBMC数据
单细胞交响乐21-实战三 批量处理并整合多个10X PBMC数据
单细胞交响乐22-实战五 CEL-seq2
单细胞交响乐23-实战六 CEL-seq
单细胞交响乐24-实战七 SMARTer 胰腺细胞
单细胞交响乐25-实战八 Smart-seq2 胰腺细胞
单细胞交响乐26-实战九 胰腺细胞数据整合
1 前言
前面的种种都是作为知识储备,但是不实战还是记不住前面的知识
这是第十个实战练习
数据来自Grun et al. 2016的小鼠造血干细胞 haematopoietic stem cell (HSC) ,使用的技术是CEL-seq
数据准备
library(scRNAseq)
sce.grun.hsc <- GrunHSCData(ensembl=TRUE)
sce.grun.hsc
# class: SingleCellExperiment
# dim: 21817 1915
# metadata(0):
# assays(1): counts
# rownames(21817): ENSMUSG00000109644
# ENSMUSG00000007777 ... ENSMUSG00000055670
# ENSMUSG00000039068
# rowData names(3): symbol chr originalName
# colnames(1915): JC4_349_HSC_FE_S13_
# JC4_350_HSC_FE_S13_ ...
# JC48P6_1203_HSC_FE_S8_
# JC48P6_1204_HSC_FE_S8_
# colData names(2): sample protocol
# reducedDimNames(0):
# altExpNames(0):
table(sce.grun.hsc$sample)
#
# JC20 JC21 JC26 JC27 JC28 JC30 JC32
# 87 96 85 91 80 96 93
# JC35 JC36 JC37 JC39 JC4 JC40 JC41
# 96 80 87 93 84 96 94
# JC43 JC44 JC45 JC46 JC48P4 JC48P6 JC48P7
# 92 94 90 96 95 96 94
ID转换
library(AnnotationHub)
ens.mm.v97 <- AnnotationHub()[["AH73905"]]
anno <- select(ens.mm.v97, keys=rownames(sce.grun.hsc),
keytype="GENEID", columns=c("SYMBOL", "SEQNAME"))
# 这里全部对应
> sum(is.na(anno$SYMBOL))
[1] 0
> sum(is.na(anno$SEQNAME))
[1] 0
# 接下来只需要匹配顺序即可
rowData(sce.grun.hsc) <- anno[match(rownames(sce.grun.hsc), anno$GENEID),]
sce.grun.hsc
## class: SingleCellExperiment
## dim: 21817 1915
## metadata(0):
## assays(1): counts
## rownames(21817): ENSMUSG00000109644 ENSMUSG00000007777 ...
## ENSMUSG00000055670 ENSMUSG00000039068
## rowData names(3): GENEID SYMBOL SEQNAME
## colnames(1915): JC4_349_HSC_FE_S13_ JC4_350_HSC_FE_S13_ ...
## JC48P6_1203_HSC_FE_S8_ JC48P6_1204_HSC_FE_S8_
## colData names(2): sample protocol
## reducedDimNames(0):
## altExpNames(0):
2 质控
依然是备份一下,把unfiltered数据主要用在质控的探索上
unfiltered <- sce.grun.hsc
发现这个数据既没有MT也没有ERCC
grep('MT',rowData(sce.grun.hsc)$SEQNAME)
# integer(0)
能用的数据只有其中的protocol了,它表示细胞提取方法
table(sce.grun.hsc$protocol)
#
# micro-dissected cells
# 1546
# sorted hematopoietic stem cells
# 369
# 再看一下各个样本与提取方法的对应关系
table(sce.grun.hsc$protocol,sce.grun.hsc$sample)

根据背景知识,大部分显微操作(micro-dissected)得到的细胞很多质量都较低,和我们的质控假设相违背,于是这里就不把它们纳入过滤条件
library(scater)
stats <- perCellQCMetrics(sce.grun.hsc)
# 只用sorted hematopoietic stem cells 计算过滤条件
qc <- quickPerCellQC(stats, batch=sce.grun.hsc$protocol,
subset=grepl("sorted", sce.grun.hsc$protocol))
colSums(as.matrix(qc))
## low_lib_size low_n_features discard
## 465 482 488
sce.grun.hsc <- sce.grun.hsc[,!qc$discard]
做个图看看
colData(unfiltered) <- cbind(colData(unfiltered), stats)
unfiltered$discard <- qc$discard
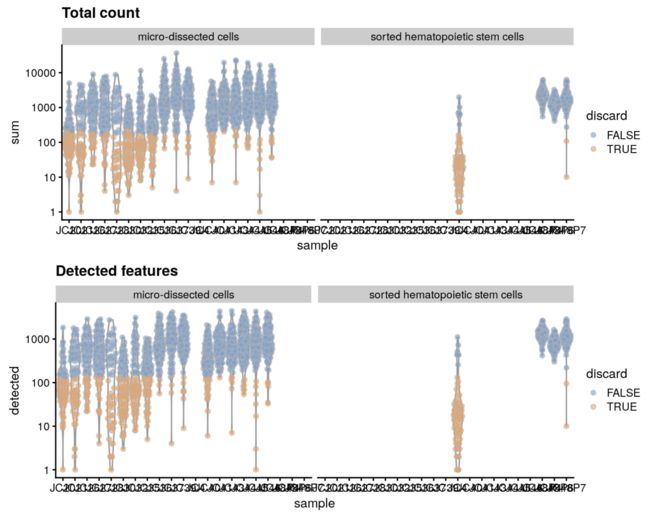
gridExtra::grid.arrange(
plotColData(unfiltered, y="sum", x="sample", colour_by="discard",
other_fields="protocol") + scale_y_log10() + ggtitle("Total count") +
facet_wrap(~protocol),
plotColData(unfiltered, y="detected", x="sample", colour_by="discard",
other_fields="protocol") + scale_y_log10() +
ggtitle("Detected features") + facet_wrap(~protocol),
ncol=1
)
可以看到,大多数的显微操作技术得到的细胞文库都比较小,相比于细胞分选方法,它在提取过程中对细胞损伤较大
3 归一化
使用去卷积方法
library(scran)
set.seed(101000110)
clusters <- quickCluster(sce.grun.hsc)
sce.grun.hsc <- computeSumFactors(sce.grun.hsc, clusters=clusters)
sce.grun.hsc <- logNormCounts(sce.grun.hsc)
4 找高变异基因
这里没有指定任何的批次,因为想保留这两种技术产生的任何差异
set.seed(00010101)
dec.grun.hsc <- modelGeneVarByPoisson(sce.grun.hsc)
top.grun.hsc <- getTopHVGs(dec.grun.hsc, prop=0.1)
做个图
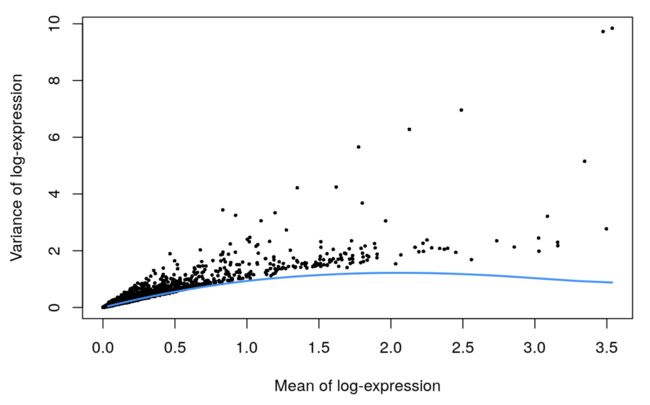
plot(dec.grun.hsc$mean, dec.grun.hsc$total, pch=16, cex=0.5,
xlab="Mean of log-expression", ylab="Variance of log-expression")
curfit <- metadata(dec.grun.hsc)
curve(curfit$trend(x), col='dodgerblue', add=TRUE, lwd=2)
看到这个线有点“太平缓”,和之前见过的都不一样,感觉“中间少了一个峰”。这是因为细胞中的基因表达量都比较低,差别也不大【大家一起贫穷,于是贫富差距很小】,所以在纵坐标(衡量变化的方差)上体现不出来差距,也就导致了拟合的曲线不会有“峰”
可能会想,那为什么不是大家表达量都很高呢(大家都很富有,贫富差距不是也很小吗)?因为横坐标可以看到,从0-3.5,这个范围对于表达量来说确实很小,之前做的图有的都大于10、15
5 降维聚类
降维就采取最基础的方式:
set.seed(101010011)
sce.grun.hsc <- denoisePCA(sce.grun.hsc, technical=dec.grun.hsc, subset.row=top.grun.hsc)
sce.grun.hsc <- runTSNE(sce.grun.hsc, dimred="PCA")
# 检查PC的数量
ncol(reducedDim(sce.grun.hsc, "PCA"))
## [1] 9
聚类
snn.gr <- buildSNNGraph(sce.grun.hsc, use.dimred="PCA")
colLabels(sce.grun.hsc) <- factor(igraph::cluster_walktrap(snn.gr)$membership)
table(colLabels(sce.grun.hsc))
##
## 1 2 3 4 5 6 7 8 9 10 11 12
## 259 148 221 103 177 108 48 122 98 63 62 18
作图
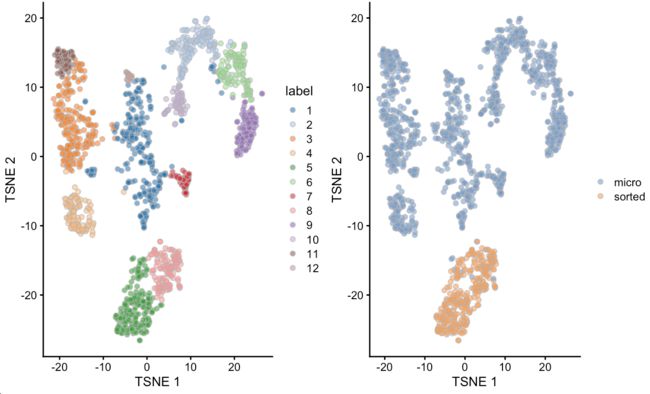
short <- ifelse(grepl("micro", sce.grun.hsc$protocol), "micro", "sorted")
gridExtra:::grid.arrange(
plotTSNE(sce.grun.hsc, colour_by="label"),
plotTSNE(sce.grun.hsc, colour_by=I(short)),
ncol=2
)
由于没有去除两个技术批次的差异,所以这里分的很开
6 找marker基因
markers <- findMarkers(sce.grun.hsc, test.type="wilcox", direction="up",
row.data=rowData(sce.grun.hsc)[,"SYMBOL",drop=FALSE])
检查一下cluster6的marker基因
chosen <- markers[['6']]
best <- chosen[chosen$Top <= 10,]
length(best)
# [1] 16
# 将cluster6与其他clusters对比的AUC结果提取出来
aucs <- getMarkerEffects(best, prefix="AUC")
rownames(aucs) <- best$SYMBOL
library(pheatmap)
pheatmap(aucs, color=viridis::plasma(100))
看到溶菌酶相关基因(LYZ家族)、Camp、 Lcn2、 Ltf 都上调,表明cluster6可能是神经元起源细胞

欢迎关注我们的公众号~_~
我们是两个农转生信的小硕,打造生信星球,想让它成为一个不拽术语、通俗易懂的生信知识平台。需要帮助或提出意见请后台留言或发送邮件到[email protected]
